Clinical
| When T is… | and N is… | and M is… | Then the Clinical Prognostic Stage Group is… |
|---|---|---|---|
| cT1 | cN0 | cM0 | I |
| cT2 | cN0 | cM0 | II |
| cT1 | cN1 | cM0 | III |
| cT2 | cN1 | cM0 | III |
| cT3 | cNX | cM0 | III |
| cT3 | cN0 | cM0 | III |
| cT3 | cN1 | cM0 | III |
| cTX | cNX | cM1 | IV |
| cTX | cNX | cM1a | IV |
| cTX | cNX | cM1b | IV |
| cTX | cNX | cM1c | IV |
| cTX | cN0 | cM1 | IV |
| cTX | cN0 | cM1a | IV |
| cTX | cN0 | cM1b | IV |
| cTX | cN0 | cM1c | IV |
| cTX | cN1 | cM1 | IV |
| cTX | cN1 | cM1a | IV |
| cTX | cN1 | cM1b | IV |
| cTX | cN1 | cM1c | IV |
| cT1 | cNX | cM1 | IV |
| cT1 | cNX | cM1a | IV |
| cT1 | cNX | cM1b | IV |
| cT1 | cNX | cM1c | IV |
| cT1 | cN0 | cM1 | IV |
| cT1 | cN0 | cM1a | IV |
| cT1 | cN0 | cM1b | IV |
| cT1 | cN0 | cM1c | IV |
| cT1 | cN1 | cM1 | IV |
| cT1 | cN1 | cM1a | IV |
| cT1 | cN1 | cM1b | IV |
| cT1 | cN1 | cM1c | IV |
| cT2 | cNX | cM1 | IV |
| cT2 | cNX | cM1a | IV |
| cT2 | cNX | cM1b | IV |
| cT2 | cNX | cM1c | IV |
| cT2 | cN0 | cM1 | IV |
| cT2 | cN0 | cM1a | IV |
| cT2 | cN0 | cM1b | IV |
| cT2 | cN0 | cM1c | IV |
| cT2 | cN1 | cM1 | IV |
| cT2 | cN1 | cM1a | IV |
| cT2 | cN1 | cM1b | IV |
| cT2 | cN1 | cM1c | IV |
| cT3 | cNX | cM1 | IV |
| cT3 | cNX | cM1a | IV |
| cT3 | cNX | cM1b | IV |
| cT3 | cNX | cM1c | IV |
| cT3 | cN0 | cM1 | IV |
| cT3 | cN0 | cM1a | IV |
| cT3 | cN0 | cM1b | IV |
| cT3 | cN0 | cM1c | IV |
| cT3 | cN1 | cM1 | IV |
| cT3 | cN1 | cM1a | IV |
| cT3 | cN1 | cM1b | IV |
| cT3 | cN1 | cM1c | IV |
| cTX | cNX | pM1 | IV |
| cTX | cNX | pM1a | IV |
| cTX | cNX | pM1b | IV |
| cTX | cNX | pM1c | IV |
| cTX | cN0 | pM1 | IV |
| cTX | cN0 | pM1a | IV |
| cTX | cN0 | pM1b | IV |
| cTX | cN0 | pM1c | IV |
| cTX | cN1 | pM1 | IV |
| cTX | cN1 | pM1a | IV |
| cTX | cN1 | pM1b | IV |
| cTX | cN1 | pM1c | IV |
| cT1 | cNX | pM1 | IV |
| cT1 | cNX | pM1a | IV |
| cT1 | cNX | pM1b | IV |
| cT1 | cNX | pM1c | IV |
| cT1 | cN0 | pM1 | IV |
| cT1 | cN0 | pM1a | IV |
| cT1 | cN0 | pM1b | IV |
| cT1 | cN0 | pM1c | IV |
| cT1 | cN1 | pM1 | IV |
| cT1 | cN1 | pM1a | IV |
| cT1 | cN1 | pM1b | IV |
| cT1 | cN1 | pM1c | IV |
| cT2 | cNX | pM1 | IV |
| cT2 | cNX | pM1a | IV |
| cT2 | cNX | pM1b | IV |
| cT2 | cNX | pM1c | IV |
| cT2 | cN0 | pM1 | IV |
| cT2 | cN0 | pM1a | IV |
| cT2 | cN0 | pM1b | IV |
| cT2 | cN0 | pM1c | IV |
| cT2 | cN1 | pM1 | IV |
| cT2 | cN1 | pM1a | IV |
| cT2 | cN1 | pM1b | IV |
| cT2 | cN1 | pM1c | IV |
| cT3 | cNX | pM1 | IV |
| cT3 | cNX | pM1a | IV |
| cT3 | cNX | pM1b | IV |
| cT3 | cNX | pM1c | IV |
| cT3 | cN0 | pM1 | IV |
| cT3 | cN0 | pM1a | IV |
| cT3 | cN0 | pM1b | IV |
| cT3 | cN0 | pM1c | IV |
| cT3 | cN1 | pM1 | IV |
| cT3 | cN1 | pM1a | IV |
| cT3 | cN1 | pM1b | IV |
| cT3 | cN1 | pM1c | IV |
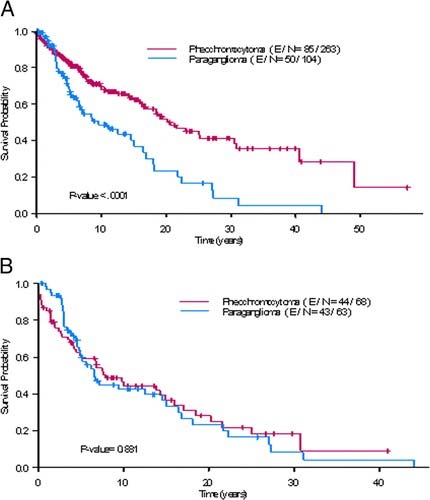
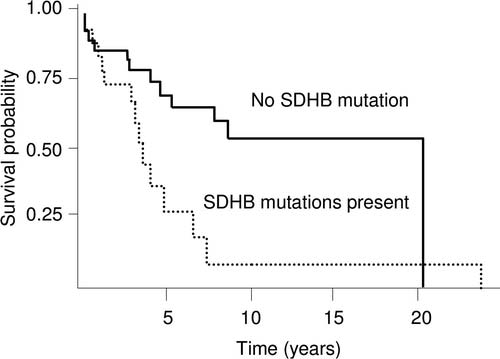
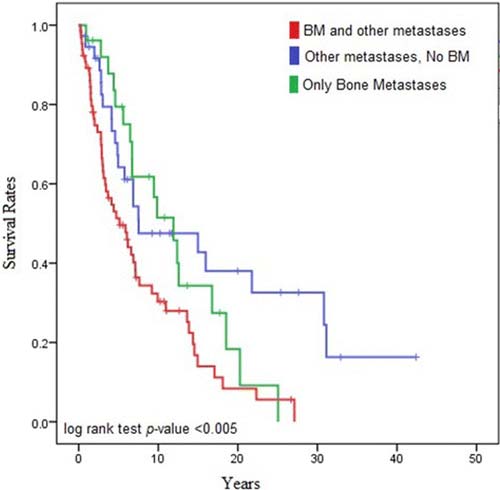
Staging applies to Pheochromocytoma/Sympathetic Paraganglioma.
Pathological
| When T is… | and N is… | and M is… | Then the Pathological Prognostic Stage Group is… |
|---|---|---|---|
| pT1 | pN0 | cM0 | I |
| pT2 | pN0 | cM0 | II |
| pT1 | pN1 | cM0 | III |
| pT2 | pN1 | cM0 | III |
| pT3 | pNX | cM0 | III |
| pT3 | pN0 | cM0 | III |
| pT3 | pN1 | cM0 | III |
| pTX | pNX | cM1 | IV |
| pTX | pNX | cM1a | IV |
| pTX | pNX | cM1b | IV |
| pTX | pNX | cM1c | IV |
| pTX | pN0 | cM1 | IV |
| pTX | pN0 | cM1a | IV |
| pTX | pN0 | cM1b | IV |
| pTX | pN0 | cM1c | IV |
| pTX | pN1 | cM1 | IV |
| pTX | pN1 | cM1a | IV |
| pTX | pN1 | cM1b | IV |
| pTX | pN1 | cM1c | IV |
| pT1 | pNX | cM1 | IV |
| pT1 | pNX | cM1a | IV |
| pT1 | pNX | cM1b | IV |
| pT1 | pNX | cM1c | IV |
| pT1 | pN0 | cM1 | IV |
| pT1 | pN0 | cM1a | IV |
| pT1 | pN0 | cM1b | IV |
| pT1 | pN0 | cM1c | IV |
| pT1 | pN1 | cM1 | IV |
| pT1 | pN1 | cM1a | IV |
| pT1 | pN1 | cM1b | IV |
| pT1 | pN1 | cM1c | IV |
| pT2 | pNX | cM1 | IV |
| pT2 | pNX | cM1a | IV |
| pT2 | pNX | cM1b | IV |
| pT2 | pNX | cM1c | IV |
| pT2 | pN0 | cM1 | IV |
| pT2 | pN0 | cM1a | IV |
| pT2 | pN0 | cM1b | IV |
| pT2 | pN0 | cM1c | IV |
| pT2 | pN1 | cM1 | IV |
| pT2 | pN1 | cM1a | IV |
| pT2 | pN1 | cM1b | IV |
| pT2 | pN1 | cM1c | IV |
| pT3 | pNX | cM1 | IV |
| pT3 | pNX | cM1a | IV |
| pT3 | pNX | cM1b | IV |
| pT3 | pNX | cM1c | IV |
| pT3 | pN0 | cM1 | IV |
| pT3 | pN0 | cM1a | IV |
| pT3 | pN0 | cM1b | IV |
| pT3 | pN0 | cM1c | IV |
| pT3 | pN1 | cM1 | IV |
| pT3 | pN1 | cM1a | IV |
| pT3 | pN1 | cM1b | IV |
| pT3 | pN1 | cM1c | IV |
| pTX | pNX | pM1 | IV |
| pTX | pNX | pM1a | IV |
| pTX | pNX | pM1b | IV |
| pTX | pNX | pM1c | IV |
| pTX | pN0 | pM1 | IV |
| pTX | pN0 | pM1a | IV |
| pTX | pN0 | pM1b | IV |
| pTX | pN0 | pM1c | IV |
| pTX | pN1 | pM1 | IV |
| pTX | pN1 | pM1a | IV |
| pTX | pN1 | pM1b | IV |
| pTX | pN1 | pM1c | IV |
| pT1 | pNX | pM1 | IV |
| pT1 | pNX | pM1a | IV |
| pT1 | pNX | pM1b | IV |
| pT1 | pNX | pM1c | IV |
| pT1 | pN0 | pM1 | IV |
| pT1 | pN0 | pM1a | IV |
| pT1 | pN0 | pM1b | IV |
| pT1 | pN0 | pM1c | IV |
| pT1 | pN1 | pM1 | IV |
| pT1 | pN1 | pM1a | IV |
| pT1 | pN1 | pM1b | IV |
| pT1 | pN1 | pM1c | IV |
| pT2 | pNX | pM1 | IV |
| pT2 | pNX | pM1a | IV |
| pT2 | pNX | pM1b | IV |
| pT2 | pNX | pM1c | IV |
| pT2 | pN0 | pM1 | IV |
| pT2 | pN0 | pM1a | IV |
| pT2 | pN0 | pM1b | IV |
| pT2 | pN0 | pM1c | IV |
| pT2 | pN1 | pM1 | IV |
| pT2 | pN1 | pM1a | IV |
| pT2 | pN1 | pM1b | IV |
| pT2 | pN1 | pM1c | IV |
| pT3 | pNX | pM1 | IV |
| pT3 | pNX | pM1a | IV |
| pT3 | pNX | pM1b | IV |
| pT3 | pNX | pM1c | IV |
| pT3 | pN0 | pM1 | IV |
| pT3 | pN0 | pM1a | IV |
| pT3 | pN0 | pM1b | IV |
| pT3 | pN0 | pM1c | IV |
| pT3 | pN1 | pM1 | IV |
| pT3 | pN1 | pM1a | IV |
| pT3 | pN1 | pM1b | IV |
| pT3 | pN1 | pM1c | IV |
| cTX | cNX | pM1 | IV |
| cTX | cNX | pM1a | IV |
| cTX | cNX | pM1b | IV |
| cTX | cNX | pM1c | IV |
| cTX | cN0 | pM1 | IV |
| cTX | cN0 | pM1a | IV |
| cTX | cN0 | pM1b | IV |
| cTX | cN0 | pM1c | IV |
| cTX | cN1 | pM1 | IV |
| cTX | cN1 | pM1a | IV |
| cTX | cN1 | pM1b | IV |
| cTX | cN1 | pM1c | IV |
| cT1 | cNX | pM1 | IV |
| cT1 | cNX | pM1a | IV |
| cT1 | cNX | pM1b | IV |
| cT1 | cNX | pM1c | IV |
| cT1 | cN0 | pM1 | IV |
| cT1 | cN0 | pM1a | IV |
| cT1 | cN0 | pM1b | IV |
| cT1 | cN0 | pM1c | IV |
| cT1 | cN1 | pM1 | IV |
| cT1 | cN1 | pM1a | IV |
| cT1 | cN1 | pM1b | IV |
| cT1 | cN1 | pM1c | IV |
| cT2 | cNX | pM1 | IV |
| cT2 | cNX | pM1a | IV |
| cT2 | cNX | pM1b | IV |
| cT2 | cNX | pM1c | IV |
| cT2 | cN0 | pM1 | IV |
| cT2 | cN0 | pM1a | IV |
| cT2 | cN0 | pM1b | IV |
| cT2 | cN0 | pM1c | IV |
| cT2 | cN1 | pM1 | IV |
| cT2 | cN1 | pM1a | IV |
| cT2 | cN1 | pM1b | IV |
| cT2 | cN1 | pM1c | IV |
| cT3 | cNX | pM1 | IV |
| cT3 | cNX | pM1a | IV |
| cT3 | cNX | pM1b | IV |
| cT3 | cNX | pM1c | IV |
| cT3 | cN0 | pM1 | IV |
| cT3 | cN0 | pM1a | IV |
| cT3 | cN0 | pM1b | IV |
| cT3 | cN0 | pM1c | IV |
| cT3 | cN1 | pM1 | IV |
| cT3 | cN1 | pM1a | IV |
| cT3 | cN1 | pM1b | IV |
| cT3 | cN1 | pM1c | IV |
Staging applies to Pheochromocytoma/Sympathetic Paraganglioma.
Neoadjuvant Clinical
There is no Postneoadjuvant Clinical Therapy (ycTNM) stage group available at this time.
Neoadjuvant Pathological
| When T is… | and N is… | and M is… | Then the Postneoadjuvant Pathological Stage Group is… |
|---|---|---|---|
| ypT1 | ypN0 | cM0 | I |
| ypT2 | ypN0 | cM0 | II |
| ypT1 | ypN1 | cM0 | III |
| ypT2 | ypN1 | cM0 | III |
| ypT3 | ypNX | cM0 | III |
| ypT3 | ypN0 | cM0 | III |
| ypT3 | ypN1 | cM0 | III |
| ypTX | ypNX | cM1 | IV |
| ypTX | ypNX | cM1a | IV |
| ypTX | ypNX | cM1b | IV |
| ypTX | ypNX | cM1c | IV |
| ypTX | ypN0 | cM1 | IV |
| ypTX | ypN0 | cM1a | IV |
| ypTX | ypN0 | cM1b | IV |
| ypTX | ypN0 | cM1c | IV |
| ypTX | ypN1 | cM1 | IV |
| ypTX | ypN1 | cM1a | IV |
| ypTX | ypN1 | cM1b | IV |
| ypTX | ypN1 | cM1c | IV |
| ypT1 | ypNX | cM1 | IV |
| ypT1 | ypNX | cM1a | IV |
| ypT1 | ypNX | cM1b | IV |
| ypT1 | ypNX | cM1c | IV |
| ypT1 | ypN0 | cM1 | IV |
| ypT1 | ypN0 | cM1a | IV |
| ypT1 | ypN0 | cM1b | IV |
| ypT1 | ypN0 | cM1c | IV |
| ypT1 | ypN1 | cM1 | IV |
| ypT1 | ypN1 | cM1a | IV |
| ypT1 | ypN1 | cM1b | IV |
| ypT1 | ypN1 | cM1c | IV |
| ypT2 | ypNX | cM1 | IV |
| ypT2 | ypNX | cM1a | IV |
| ypT2 | ypNX | cM1b | IV |
| ypT2 | ypNX | cM1c | IV |
| ypT2 | ypN0 | cM1 | IV |
| ypT2 | ypN0 | cM1a | IV |
| ypT2 | ypN0 | cM1b | IV |
| ypT2 | ypN0 | cM1c | IV |
| ypT2 | ypN1 | cM1 | IV |
| ypT2 | ypN1 | cM1a | IV |
| ypT2 | ypN1 | cM1b | IV |
| ypT2 | ypN1 | cM1c | IV |
| ypT3 | ypNX | cM1 | IV |
| ypT3 | ypNX | cM1a | IV |
| ypT3 | ypNX | cM1b | IV |
| ypT3 | ypNX | cM1c | IV |
| ypT3 | ypN0 | cM1 | IV |
| ypT3 | ypN0 | cM1a | IV |
| ypT3 | ypN0 | cM1b | IV |
| ypT3 | ypN0 | cM1c | IV |
| ypT3 | ypN1 | cM1 | IV |
| ypT3 | ypN1 | cM1a | IV |
| ypT3 | ypN1 | cM1b | IV |
| ypT3 | ypN1 | cM1c | IV |
| ypTX | ypNX | pM1 | IV |
| ypTX | ypNX | pM1a | IV |
| ypTX | ypNX | pM1b | IV |
| ypTX | ypNX | pM1c | IV |
| ypTX | ypN0 | pM1 | IV |
| ypTX | ypN0 | pM1a | IV |
| ypTX | ypN0 | pM1b | IV |
| ypTX | ypN0 | pM1c | IV |
| ypTX | ypN1 | pM1 | IV |
| ypTX | ypN1 | pM1a | IV |
| ypTX | ypN1 | pM1b | IV |
| ypTX | ypN1 | pM1c | IV |
| ypT1 | ypNX | pM1 | IV |
| ypT1 | ypNX | pM1a | IV |
| ypT1 | ypNX | pM1b | IV |
| ypT1 | ypNX | pM1c | IV |
| ypT1 | ypN0 | pM1 | IV |
| ypT1 | ypN0 | pM1a | IV |
| ypT1 | ypN0 | pM1b | IV |
| ypT1 | ypN0 | pM1c | IV |
| ypT1 | ypN1 | pM1 | IV |
| ypT1 | ypN1 | pM1a | IV |
| ypT1 | ypN1 | pM1b | IV |
| ypT1 | ypN1 | pM1c | IV |
| ypT2 | ypNX | pM1 | IV |
| ypT2 | ypNX | pM1a | IV |
| ypT2 | ypNX | pM1b | IV |
| ypT2 | ypNX | pM1c | IV |
| ypT2 | ypN0 | pM1 | IV |
| ypT2 | ypN0 | pM1a | IV |
| ypT2 | ypN0 | pM1b | IV |
| ypT2 | ypN0 | pM1c | IV |
| ypT2 | ypN1 | pM1 | IV |
| ypT2 | ypN1 | pM1a | IV |
| ypT2 | ypN1 | pM1b | IV |
| ypT2 | ypN1 | pM1c | IV |
| ypT3 | ypNX | pM1 | IV |
| ypT3 | ypNX | pM1a | IV |
| ypT3 | ypNX | pM1b | IV |
| ypT3 | ypNX | pM1c | IV |
| ypT3 | ypN0 | pM1 | IV |
| ypT3 | ypN0 | pM1a | IV |
| ypT3 | ypN0 | pM1b | IV |
| ypT3 | ypN0 | pM1c | IV |
| ypT3 | ypN1 | pM1 | IV |
| ypT3 | ypN1 | pM1a | IV |
| ypT3 | ypN1 | pM1b | IV |
| ypT3 | ypN1 | pM1c | IV |
Staging applies to Pheochromocytoma/Sympathetic Paraganglioma.