Pancreatic neuroendocrine tumors (NETs) exhibit neuroendocrine differentiation and comprise less than 2% of all pancreatic malignancies. Although these tumors are rare, their relatively indolent nature translates into a relatively high prevalence: approximately 10% of all pancreatic tumors.1 An analysis of the Surveillance, Epidemiology, and End Results (SEER) database, 1973 to 2004, suggests that the incidence of pancreatic NETs is increasing.2 In 1973, the age-adjusted incidence of pancreatic NETs in the US population was 0.18 per 100,000; in 2003, it was 0.30 per 100,000.2 A recent analysis of the national population-based Cancer Registry of Norway revealed a similar trend.3 The age-stand ardized incidence rate of pancreatic NETs in the population overall (1993-2010) was 0.47 per 100,000 (95% CI, 0.43-0.52); in 2006 to 2010, it was 0.71 per 100,000 (95% CI, 0.61-0.82). The estimated annual percentage change was +6.9%. The reason for the increasing incidence likely is multifactorial; it probably is a result, at least in part, of more accurate classification by pathologists and improved diagnostic tools (cross-sectional and functional imaging), the latter of which has led to an increase in incidentally discovered tumors.4 Pancreatic NETs appear to be slightly more common in men (53%).1 They may occur at any age but are most commonly detected in the fifth to eighth decades; the median age at diagnosis is 60.2 With the exception of patients with insulinoma, patients with pancreatic NETs often present with advanced disease.2,5 Increased detection of incidental tumors has led to a reduction in the proportion of patients diagnosed with metastatic disease at presentation.6 Autopsy studies assessing the presence of small (less than 1 cm) NETs reported frequencies ranging from 0.8-10%.7
The grading classification scheme for pancreatic NETs has evolved over the years to encompass all NETs arising in the pancreas and gastrointestinal tract. Developed by the European Neuroendocrine Tumor Society (ENETS) and adopted by the World Health Organization (WHO) in 2010, the most common classification system consists of three grades (G1, G2, and G3), which correspond to well-differentiated (G1 and G2) and poorly differentiated neoplasms (G3).8-11
Grade is a significant and independent predictor of outcome.12,13 Most NETs arising in the pancreas are well differentiated (G1 and G2) tumors, which are termed pancreatic neuroendocrine tumors, pancreatic NETs, or panNETs. Poorly differentiated neoplasms are termed pancreatic neuroendocrine carcinomas or pancreatic NECs. Although this classification implies that all high-grade tumors are poorly differentiated, recent data suggest that a significant fraction of patients with well-differentiated tumors have a ki-67 index greater than 20%, and usually less than 50%. These “well-differentiated, high-grade” tumors represent a favorable prognostic category compared with poorly differentiated NECs.14
Approximately 20% of pancreatic NETs are associated with a clinical syndrome due to hormone excess. These “functional” tumors (F-pancreatic NETs) thus are defined based on the clinical syndrome, as asymptomatic production of hormones also may be detected in nonfunctional tumors (NF-pancreatic NETs).15 Among functional tumors, the most common hormones produced are insulin and gastrin (Table 34.1).16 Overproduction of glucagon, vasoactive intestinal peptide, or proinsulin is less common.17 Other, rarer hormone-mediated syndromes also have been reported, including pancreatic NETs secreting adrenocorticotropic hormone (ACTH), leading to Cushing's syndrome (ACTHomas); pancreatic NETs causing the carcinoid syndrome; and pancreatic NETs causing hypercalcemia (PTHrp-omas).18,19 Pancreatic NETs associated with calcitonin or ACTH production appear to be relatively aggressive, as do those that switch from one functional syndrome to another.
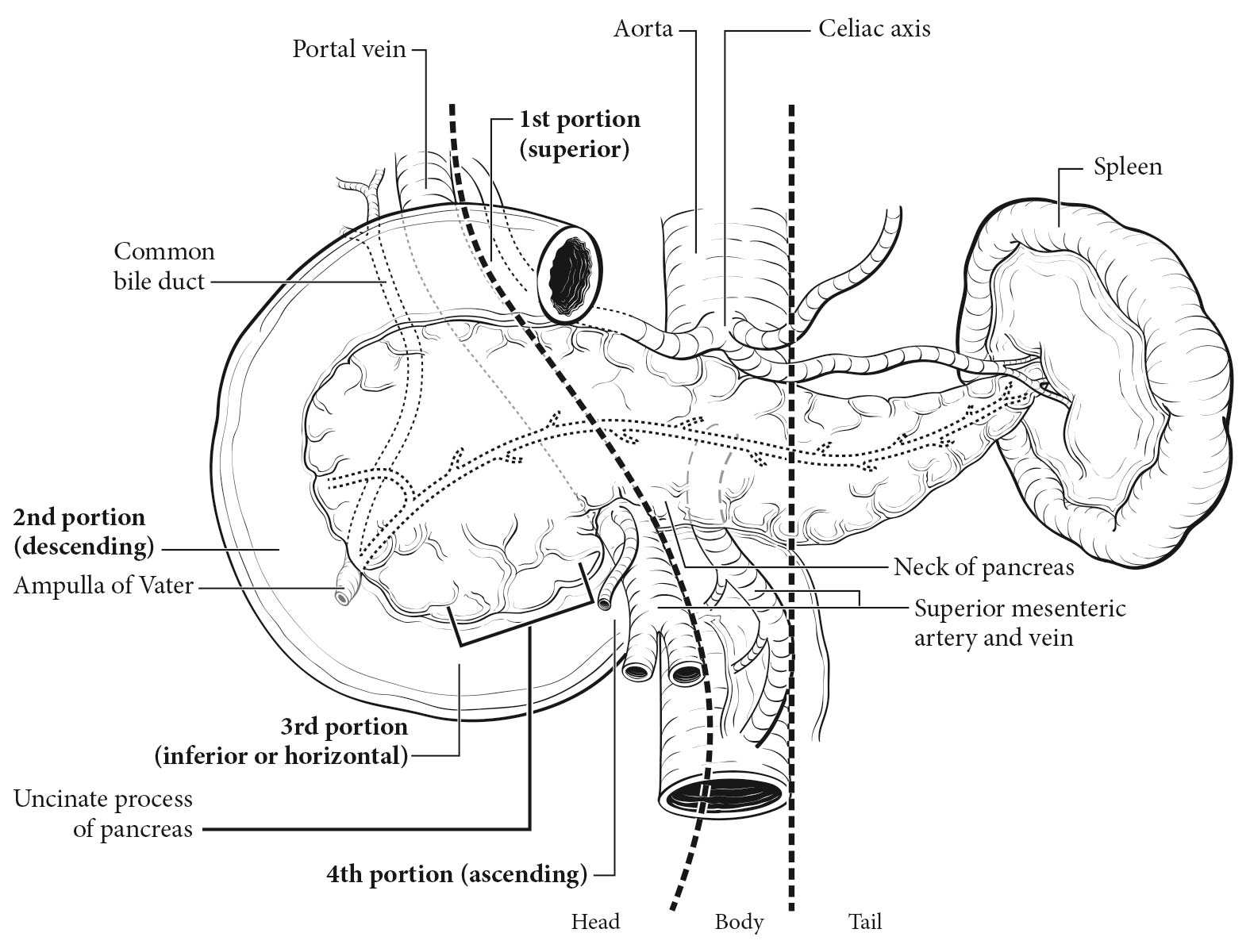
More than 50% of functional tumors are located in the tail of the pancreas, the exception being gastrinomas, which are more likely (63%) to be located in the head of the pancreas (Table
34.1).
1 Interestingly, most gastrinomas (60-80%) actually arise in the duodenum; 75-85% are located in the “gastrinoma triangle” involving the duodenum and pancreatic head.
17 Insulinomas typically are small, well-circumscribed tumors that are diagnosed at an early stage as the result of symptoms associated with hypoglycemia. The vast majority do not recur after resection. Importantly, F-pancreatic NETs may produce more than one hormone, and hormone production may change over the course of tumor progression.
20
34.1 Clinical features of functional pancreatic neuroendocrine tumors
| Name | Biologically active peptide(s) | Incidence(new cases/106 population/year) | Tumor location | Most common symptoms/signs |
|---|
| Most common syndromes |
| Insulinoma | Insulin | 1-3 | Pancreas (greater than 99%) | Hypoglycemic symptoms (Whipple's triad) |
| Zollinger-Ellison syndrome | Gastrin | 0.5-2 | Duodenum (70%); pancreas (25%); other sites (5%) | Abdominal pain, gastroesophageal reflux, diarrhea, duodenal ulcers, PUD/GERD |
| Less common syndromes (additional, rarer syndromes also exist) |
| VIPoma (Verner-Morrison syndrome, pancreatic cholera, WDHA syndrome) | Vasoactive intestinal peptide | 0.05-0.2 | Pancreas (90%, adult); other (10%, neural, adrenal, periganglionic) | Diarrhea, hypokalemia, dehydration |
| Glucagonoma | Glucagon | 0.01-0.1 | Pancreas (100%) | Rash, glucose intolerance, weight loss |
| Somatostatinoma | Somatostatin | Rare | Pancreas (55%); duodenum/jejunum (44%) | Diabetes mellitus, cholelithiasis, diarrhea |
| ACTHoma | ACTH | Rare | Pancreas (4-16% all ectopic Cushing's) | Cushing's syndrome |
| Pancreatic NET causing carcinoid syndrome | Serotonin | Rare | Pancreas (less than 1% all carcinoid syndrome) | Flushing, diarrhea |
| PTHrp-oma (hypercalcemia) | PTHrp, others unknown | Rare | Pancreas | Symptoms due to hypercalcemia |
| Abbreviations: GERD, gastroesophageal reflux disease; PTHrp, parathyroid hormone-related protein; PUD, peptic ulcer disease; WDHA, watery diarrhea, hypokalemia, and achlorhydria. (Adapted from Jensen et al.17) |
Pancreatic NETs frequently secrete several substances into the serum, including chromogranin A (CgA), pancreatic polypeptide (PP), pancreastatin, and neuron-specific enolase, without obvious clinical consequence. As such (assuming they are not secreting any additional hormones, as listed in Table 34.1), these tumors are considered NF-pancreatic NETs.
18,
21-
23 NF-pancreatic NETs occur at least twice as frequently as F-pancreatic NETs in most series.
The etiology of pancreatic NETs largely is unknown. Most pancreatic NETs are thought to be sporadic. Of these, approximately 43% harbor DAXX/ATRX mutations, 44% harbor somatic inactivating mutations of MEN1, and 15% contain mutations in genes encoding mTOR pathway components.24 The prognostic significance of these mutations remains to be determined definitively.24,25 A small fraction of pancreatic NETs (less than 10%) arise in the context of a hereditary cancer syndrome, the most common of which is multiple endocrine neoplasia type 1 (MEN1).26 MEN1 is caused by mutations in the MEN1 gene located at chromosome 11q13 region, thus altering transcriptional regulation, genomic stability, cell division, and cell cycle control.27 Affected patients develop hyperplasia or neoplasia of multiple endocrine and nonendocrine tissues, including parathyroid adenomas (95-100%) resulting in hyperparathyroidism, pituitary adenomas (54-65%), adrenal adenomas (27-36%), various NETs (gastric, lung, thymic; 0-10%), thyroid adenomas (up to 10%), various skin tumors (80-95%), central nervous system tumors (up to 8%), and smooth muscle tumors (up to 10%).17,28 Pancreatic NETs develop in 80-100% of MEN1 patients and are nearly always multifocal. Pancreatic NETs in this setting often are small and nonfunctional. Gastrinomas (greater than 80% duodenal) develop in 54% of MEN1 patients, insulinomas in 18%, and glucagonomas, vasoactive intestinal peptide-secreting tumors (VIPomas), growth hormone-releasing factor-secreting tumors (GRFomas), and somatostatinomas in less than 5%.17 Pancreatic NETs also occur in up to 10% of patients with von Recklinghausen's disease (also known as neurofibromatosis type 1 [NF1]), 10-17% of patients with von Hippel-Lindau (VHL) syndrome, and rarely in patients with tuberous sclerosis. The likelihood of an underlying genetic syndrome depends on the type of tumor as well as the patient's personal and family history, which should be recorded for every patient presenting with a pancreatic NET. Multifocal disease is a risk factor, as is the type of hormone produced. MEN1 is present in 20-30% of patients with Zollinger-Ellison syndrome (ZES; usually associated with a duodenal gastrinoma) and 5% of patients with an insulinoma.17 Given the high incidence of parathyroid adenomas in MEN1 patients, assessment of ionized calcium and serum parathyroid hormone (PTH) may be used as a screening tool in appropriate patients, with the caveat that secondary elevation of PTH may occur in the setting of vitamin D deficiency.29,30
The staging of pancreatic NETs depends on the size and extent of the primary tumor (including whether there is lymph node involvement and /or distant metastasis). Importantly, the American Joint Committee on Cancer (AJCC) TNM classification system for pancreatic NETs was first introduced in the AJCC Cancer Staging Manual, 7th Edition and was based on the staging algorithm for exocrine pancreatic carcinomas. Recognizing that exocrine and neuroendocrine tumors of the pancreas are distinct entities in terms of underlying tumor biology and prognosis, other staging classification systems have been proposed.12,13,31-33 The 7th Edition AJCC staging system is significantly associated with survival, but its value is limited by the inability to discriminate between the intermediate stages (i.e., Stages II and III are prognostically indistinguishable).12 Furthermore, some of the parameters necessary for staging (e.g., presence of extrapancreatic extension) are difficult to assess pathologically because of the expansile growth pattern common in pancreatic NETs. The system developed by ENETS in 2006 incorporates a narrower T definition. It has proven prognostic value and appears to provide superior distinction among stages (I, II, III, and IV) compared with the AJCC/Union for International Cancer Control (UICC)/WHO 2010 system.12 Therefore, the AJCC Cancer Staging Manual, 8th Edition staging system has been modified to be consistent with the ENETS system.
Importantly, even the current ENETS staging system is imperfect; patients with Stage IIIB (any T N1 M0) disease fare better than those with Stage IIIA (T4 N0 M0) disease, and clear discrimination between stages has not been evident in all validation studies.12,13 There are several potential reasons for this, including the generally favorable survival outcomes of patients with nonmetastatic disease, which limits the ability to distinguish prognostic groups, especially in the absence of very long-term follow-up. In addition, inconsistent lymph node sampling may underlie the conflicting findings of the prognostic importance of lymph node involvement.34 Further refinement of the ENETS staging system is likely, although inadequate data exist for modifications at present.12,33
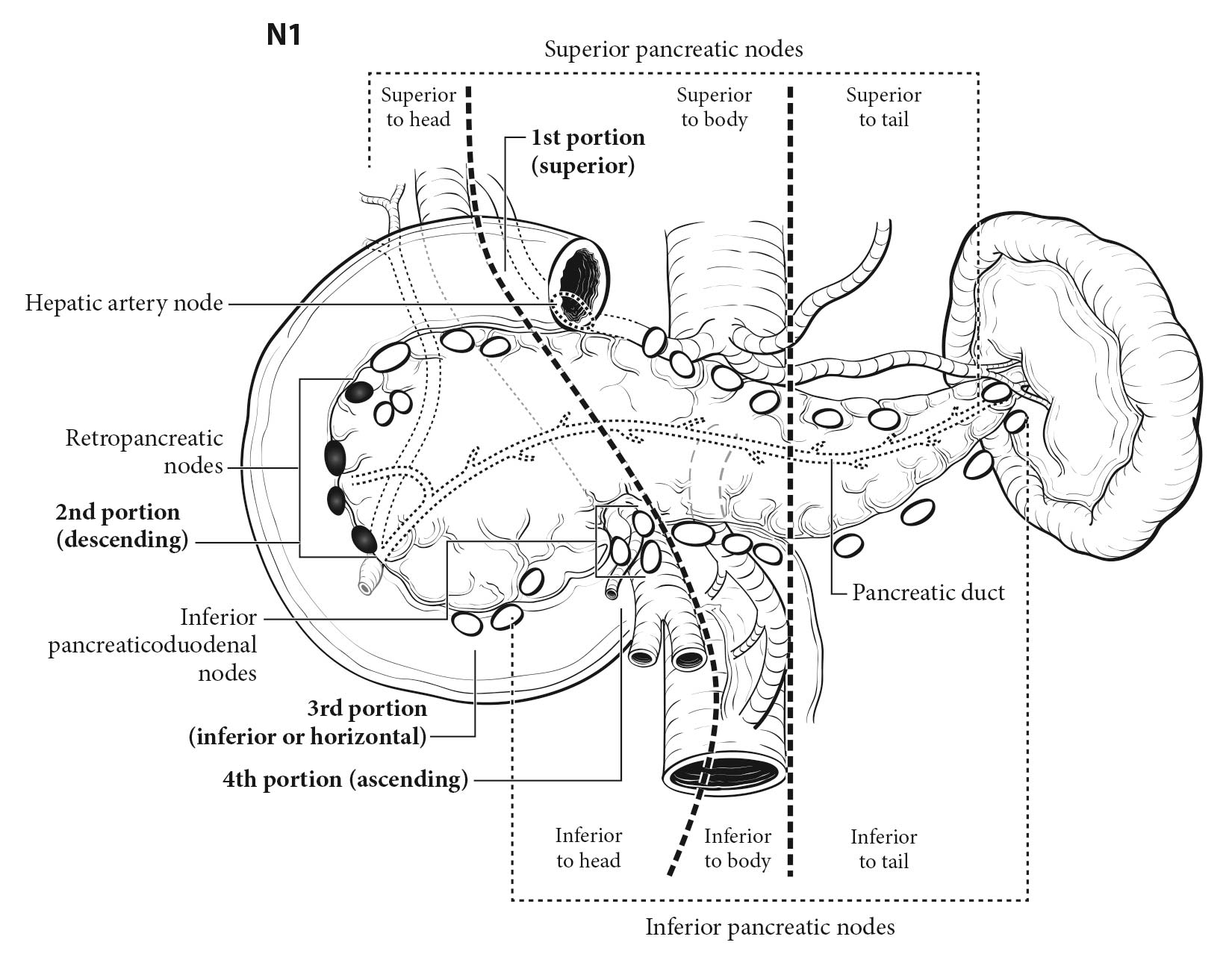
Surgical resection remains the only potentially curative treatment for well-differentiated (G1/G2) pancreatic NETs. The natural history of these tumors is poorly understood because of their relative rarity, but accepted prognostic factors include patient age, distant metastases, tumor grade, and tumor differentiation.12 Recent studies suggest that lymph node involvement also may be an important prognostic factor.35,36 For accurate staging, routine lymph node sampling is critical for most patients with pancreatic NETs undergoing surgery.
Well-differentiated insulinomas rarely metastasize and have a particularly good prognosis (greater than 90% have a benign clinical course); the prognosis of other functional tumors appears to match that of nonfunctional tumors in most series, although this is not consistent in all studies.12 The type of surgery performed depends on the tumor stage, location, and functional status, and ranges from enucleation to pancreaticoduodenectomy.37 Because insulinomas typically are small and pursue a benign clinical course, enucleation of tumors located away from the main pancreatic duct usually is curative. For insulinomas close to the main pancreatic duct, a pancreatectomy, such as distal pancreatectomy for left-sided lesions or pancreaticoduodenectomy for right-sided lesions, may be required. Insulinomas located in the pancreatic neck region may be treated with a central pancreatectomy. Because most insulinomas are very indolent, a lymphadenectomy typically is not necessary, and spleen preservation may be considered.
In contrast, most NF-pancreatic NETs and other F-pancreatic NETs (i.e., not insulinomas) are capable of malignant behavior. The optimal treatment of incidentally identified small NF-pancreatic NETs less than 1.5 cm is unclear. Recent analyses suggest that surveillance, rather than surgery, is appropriate in many cases. A careful analysis of the potential risks and benefits of pancreatic resection is required, particularly in asymptomatic elderly patients with significant comorbidities.4,38
Most larger NF-NETs or localized pancreatic NETs with an elevated proliferative index have a higher risk of invasion and metastases; thus, resection with a lymphadenectomy should be strongly considered for these neoplasms. Even small pancreatic NETs may be associated with significant lymph node and /or liver metastases.39 Several factors influence the choice of surgical procedure, including primary tumor size, Ki-67 labeling index, mitotic index, location, and medical comorbidities. For left-sided lesions, a distal pancreatectomy and , if necessary, an en bloc splenectomy should be done to ensure adequate lymphadenectomy. A pancreaticoduodenectomy (Whipple procedure) should be considered for right-sided lesions. Rarely, a total pancreatectomy with en bloc splenectomy is required for large lesions that occupy much of the pancreas, but given the high morbidity of such a procedure, a thorough evaluation should be performed first to rule out metastatic disease. A central pancreatectomy with regional lymphadenectomy might be considered for lesions located in the pancreatic neck. If enucleation is considered for these neoplasms, a regional lymphadenectomy may be done to ensure adequate lymph node sampling for staging. Removal of a pancreatic primary tumor in the setting of resectable liver metastases may be considered, particularly if a pancreaticoduodenectomy is not required. Although complete resection and /or palliative debulking surgeries (i.e., primary tumor and liver metastases) are not necessarily curative, data from nonrand omized studies suggest they may be associated with improved hormone-mediated symptoms and improved survival in carefully selected patients.40,41
Several treatment options exist for patients with advanced, unresectable pancreatic NETs. Somatostatin analogs have cytostatic activity and may be used to treat hormone-mediated symptoms.42-44 Chemotherapy has been used with some success, although the optimal regimen remains unclear.45-47 Two targeted therapies are approved for this indication: everolimus (an inhibitor of mTOR signaling) and sunitinib (an oral inhibitor of vascular endothelial growth factor signaling) both delay progression of progressive panNETs.48,49 The use of liver-directed therapy or other treatments depends on several factors, including the tumor's growth rate, extent of disease, and whether the tumor is functional. See published guidelines for additional information regarding the workup and treatment of panNETs.17,37,50