AUTHORS: David Qu, MD and Anna Yang, MD


DefinitionNeurofibromatosis (NF) is an autosomal-dominant tumor predisposition syndrome affecting bone, nervous system, soft tissue, and skin. Traditionally NF has been divided into three major subtypes: NF type 1 (NF1), NF type 2 (NF2), and schwannomatosis (SWN). However, nomenclature and diagnostic criteria for neurofibromatosis were recently updated by an international panel of experts in 2022 to incorporate clinical and genetic discoveries made since the previous consensus conference in the late 1990s. This was released just 3 months prior to updating this chapter, so although these changes are included here, older literature based on the prior nomenclature and diagnostic criteria is still part of this chapter as well.
SynonymsNF1: NF, von Recklinghausen disease, peripheral NF
Schwannomatosis, as further subdivided:
- NF2-related schwannomatosis (previous known as NF2 or central NF)
- SMARCB1-related schwannomatosis
- LZTR1-related schwannomatosis
- 22q-related schwannomatosis
- Schwannomatosis-not otherwise specified (NOS)
- Schwannomatosis-not elsewhere classified (NEC)
- NF
| ICD-10CM CODES | | Q85.00 | Neurofibromatosis, unspecified | | Q85.01 | Neurofibromatosis, type 1 | | Q85.02 | Neurofibromatosis, type 2 | | Q85.03 | Schwannomatosis |
|
∗As of August 2022, ICD-10 codes still reflect previous classification of neurofibromatosis
Epidemiology & Demographics
- NF1 and NF2-related SWN are autosomal dominant; approximately 50% of cases have no family history.
- NF1 has an incidence of 1/3000 live births and prevalence of 1/5000 persons.
- NF2-related SWN has an incidence of 1/25,000 live births and prevalence of 1/60,000 persons.
- These two disorders affect approximately 100,000 people in the US.
- Affects males and females equally.1
- NF1 may be associated with optic gliomas, astrocytomas, spinal neurofibromas, pheochromocytomas, and chronic myeloid leukemia.
- NF2-related SWN may be associated with acoustic neuromas, meningiomas, spinal schwannomas, and gliomas.2
- The incidence of non-NF2-related SWN is 1/40,000 live births. The disease is mostly sporadic in nature; however, approximately 20% of cases appear to be inherited.1
Physical Findings & Clinical Presentation
- Common features of NF1 include:
- Café-au-lait macules (100% of children by age 2 yr)
- Hyperpigmented skin lesions (Fig. E1) occurring anywhere on the body except the face, palms, and soles
- Appear early in life and increase in size and number during puberty
- Are focal or diffuse
- Axillary and inguinal freckling (70%)
- Multiple neurofibromas (Fig. E2) can be soft or firm; three subtypes:
- Cutaneous: Circumscribed, not specific for NF1
- Subcutaneous: Circumscribed, not specific for NF1
- Plexiform: Noncircumscribed, thick and irregular; can cause disfigurement of supportive structures and is specific for NF1
- Lisch nodule (small hamartoma of the iris) found in >90% of adult cases
- Visual defects possibly related to optic gliomas (2% to 5%)
- Neurodevelopment problems, such as learning disability and mental retardation (30% to 40%)
- Skeletal disorders, including long bone dysplasia, pseudoarthrosis, scoliosis, short stature, and decreased bone mineral density.3
- Common features of NF2-related SWN include:
- Hearing loss and tinnitus related to bilateral acoustic neuromas (>90% of adults)
- Cataracts (81%), retinal hamartoma, and epiretinal membranes
- Headache, which may be due to intracranial meningiomas, present in 80% of patients
- Unsteady gait
- Cutaneous and subcutaneous neurofibromas but fewer than in NF1
- Café-au-lait macules (1%) (Fig. E3).3,4
- Common features of SMARCB1-, LZTR1-, and 22q-related SWN include:5
- Painful multiple schwannomas of the spinal (74%), peripheral (89%), or cranial nerves except the vestibular nerve (9%).
Figure E1 Systemic features of neurofibromatosis type 1.
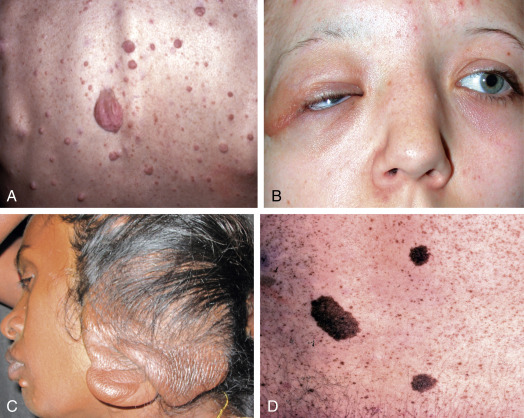
(A) Discrete neurofibromas. (B) Nodular plexiform neurofibroma of the eyelid. (C) Elephantiasis nervosa. (D) Café-au-lait spots.
Courtesy S. Kumar Puri. From Kanski JJ, Bowling B: Clinical ophthalmology, a systematic approach, ed 7, Philadelphia, 2010, Saunders.
Figure E2 Multiple neurofibromas are present in this individual.
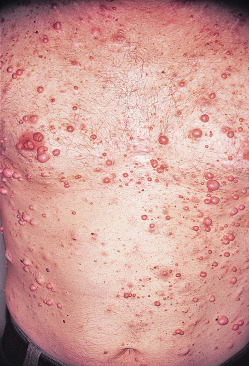
From Callen JP et al: Dermatological signs of systemic disease, ed 5, Philadelphia, 2017, Elsevier, pp 345-358.
Figure E3 A Café-Au-Lait Spot and Multiple Freckles (Crowe Sign) in the Axillary Vault is Seen in This Patient with Neurofibromatosis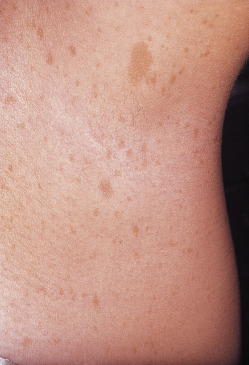
From Callen JP et al: Dermatological signs of systemic disease, ed 5, Philadelphia, 2017, Elsevier, pp 345-358.
Etiology
- NF1 is caused by DNA mutations located at 17q11.2 on chromosome 17, which is responsible for encoding the protein neurofibromin, a guanosine triphosphatase protein modulating signaling through the Ras/MAPK and PI3K/mTOR pathways.
- NF2-related SWN is caused by DNA mutations located at 22q12.2 on chromosome 22 and is responsible for encoding the protein merlin, a potent inhibitor of glioma growth that modulates signaling through the PI3K/AKT, Raf/MEK/ERK, and mTOR pathways.
- Both proteins are speculated to act as tumor suppressors.1
- Over 50% de novo NF2-related SWN have mosaicism.4
- Germline mutations in tumor suppressor genes SMARCB1 and LZTR1 on chromosome 22 account for 70% to 80% of non-NF2-related familial SWN. However, additional somatic mutations are required to trigger this condition. A 4-hit hypothesis has been proposed for tumorigenesis in SMARCB1- and LZTR1-related SWN.

- The major clinical features of NF1 are summarized in Table E1.6 Diagnostic criteria, as proposed by an international consensus released by the Children’s Tumor Foundation in 2021, are listed below. NF1 is diagnosed if the person has two or more of the seven following features:
- Six or more café-au-lait macules >5 mm in prepubertal patients and >15 mm in post-pubertal patients
- Two or more neurofibromas of any type or one plexiform neurofibroma
- Axillary or inguinal freckling
- Optic glioma
- Two or more Lisch nodules (iris hamartomas)
- A distinctive osseous lesion such as sphenoid wing dysplasia, anterolateral bowing of the tibia, or pseudarthrosis of a long bone
- A heterozygous pathogenic NF1 variant with a variant allele fraction of 50% in apparently normal tissue such as white blood cells
Additionally, a child of a parent who meets the diagnostic criteria specified above merits a diagnosis of NF1 if one or more criteria listed above are present.7
- Diagnostic criteria for both NF2 and SWN were restructured by an international consensus released by the Children’s Tumor Foundation in June 2022 in Genetics in Medicine after it was revealed that a significant number of patients who were clinically diagnosed with one (NF2 or SWN) actually have the other on genetic analysis. Instead of being a separate entity, NF2 has been found to be part of the spectrum of schwannomatosis and is now termed NF2-related schwannomatosis, which can be diagnosed by any of the following criteria:
- Bilateral vestibular schwannomas
- An identical NF2 pathogenic variant in at least two anatomically distinct NF2-related tumors (schwannoma, meningioma, and/or ependymoma)
- Either two major or one major and two minor criteria as described below:
- Major criteria
- Unilateral vestibular schwannoma
- First-degree relative other than sibling with NF2-related schwannomatosis
- Two or more meningiomas (single meningioma qualifies as minor criteria)
- NF2 pathogenic variant in an unaffected tissue such as blood
- Minor criteria
- Can count >one of a type (e.g., two distinct schwannomas would count as two minor criteria): ependymoma, schwannoma, meningioma (multiple meningiomas qualify as a major criteria)
- Can count only once (e.g., bilateral cortical cataracts count as a single minor criterion): meningioma, juvenile subcapsular or cortical cataract, retinal hamartoma, epiretinal membrane in a person <40 yr.4
- As stated previously, diagnostic criteria for SWN were updated in 2022. SWN is now subdivided into NF2-related schwannomatosis (see diagnostic criteria above), SMARCB1-related schwannomatosis, LZTR1-related schwannomatosis, 22q-related schwannomatosis, schwannomatosis NOS (not otherwise specified), and schwannomatosis NEC (not elsewhere classified).
- SMARCB1- and LZTR1-related schwannomatosis are diagnosed if the person has any of the following criteria:
- At least one pathologically confirmed schwannoma or hybrid nerve sheath tumor and a SMARCB1 (or LZTR1) pathogenic variant in an unaffected tissue such as blood
- A shared SMARCB1 (or LZTR1) pathogenic variant in two schwannomas or hybrid nerve sheath tumors.
- 22q-related schwannomatosis diagnosis can be made if the person does not meet criteria for NF2-, SMARCB1-, or LZTR1-related schwannomatosis, does not have a germline DGCR8 pathogenic variant, and has both LOH of the same chromosome 22q markers in two anatomically distinct schwannomas or hybrid nerve sheath tumors and a different NF2 pathogenic variant in each tumor which cannot be detected in unaffected tissue.
- Schwannomatosis-NOS is reserved for patients who have clinical features of NF2/SWN but have not had molecular analysis.
- Schwannomatosis-NEC is reserved for patients in whom molecular analysis of blood and tumors has failed to detect a pathogenic variant (PV).4
TABLE E1 Major Clinical Features of Neurofibromatosis Type 1
| Cutaneous |
- Neurofibromas (60%-90%)
- Café-au-lait macules (>90%)
- Axillary and/or inguinal freckling (∼80%)
- Plexiform neurofibroma (25%)
- Nevus anemicus (30%-50%)
- Juvenile xanthogranuloma (15%-35% in first 3 yr of life)
|
| Ocular |
- Lisch nodules (∼90% by 20 yr of age)
- Choroidal nodules (>80% of adults)
- Neovascular glaucoma, retinal vasoproliferative tumors
|
| Skeletal |
| Cranial |
- Macrocephaly (20%-50%)
- Hypertelorism (25%)
- Sphenoid wing dysplasia (<5%)
|
| Spinal |
- Scoliosis (5%-10%)
- Spina bifida
|
| Limbs |
- Dysplasia of long bone cortex (5%), pseudarthrosis (2%)
|
| Other |
- Generalized osteopenia (∼50%), osteoporosis (∼20%)
- Short stature (∼30% <3rd percentile)
- Pectus deformity (30%-50%)
|
| Tumors |
- Optic glioma (10%-15%) (with or without precocious puberty)
- Malignant peripheral nerve sheath tumors (3%-15%)
- Pheochromocytoma (∼1%)
- Juvenile myelomonocytic leukemia
- CNS tumors other than optic gliomas (∼5%)
- Rhabdomyosarcoma, especially of the genitourinary tract
- Duodenal carcinoid
- Somatostatinoma
- Parathyroid adenoma
- Gastrointestinal stromal tumors (GIST)
- Breast cancer (∼5-fold increased risk in women <50 yr of age)
|
| Neurologic |
- Unidentified bright objects (UBO) on MRI (50%-75%)
- Learning difficulties (30%-50%)
- Seizures (∼5%)
- Intellectual impairment (severe in <5%)
- Aqueductal stenosis and hydrocephalus (∼2%)
|
| Cardiovascular |
- Hypertension (∼30%): Essential >renal artery stenosis or pheochromocytoma
- Pulmonic stenosis (∼1%)
- Renal artery stenosis (∼2%)
- Cerebrovascular anomalies (2%-5%), including vascular stenoses and aneurysms
|
JXGs, Juvenile xanthogranulomas; MRI, magnetic resonance imaging.
From Bolognia JL: Dermatology, ed 4, Philadelphia, 2018, Elsevier, pp 985-1003.
Differential Diagnosis
- Abdominal NF
- Myxoid lipoma
- Nodular fasciitis
- Fibrous histiocytoma
- Segmental NF
- Metastasis of other tumors
- Genetic syndromes associated with nervous system tumors (Table E2)
TABLE E2 Genetic Syndromes Associated With Nervous System Tumors
| Syndrome | Chromosome or Gene | Inheritance | CNS Tumors |
|---|
| Neurofibromatosis type 1 (NF1) | 17q1 | Autosomal dominant | Pilocytic astrocytomas of the optic nerve, other gliomas, meningiomas, nerve sheath tumors |
| NF2 | 22q | Autosomal dominant | Nerve sheath tumors, glioma, meningioma |
| Li-Fraumeni syndrome | TP53 germline mutations | Autosomal dominant | Glioma, medulloblastoma, choroid plexus tumors, nerve sheath tumors, meningioma |
| Turcot syndrome | APC and hMLH1/hPSM2 germline mutations | Autosomal dominant | Medulloblastoma |
| Gorlin syndrome | 9q22.3 microdeletions
PTCH1 gene | Autosomal dominant | Medulloblastoma |
| Tuberous sclerosis | 9q32-34 | Autosomal dominant | Subependymal giant cell astrocytoma (SEGA) |
| Von Hippel-Lindau disease | 3p13-14 | Autosomal dominant | Hemangioblastoma |
| 3p25-26 |
APC, Adenomatous polyposis coli; CNS, central nervous system.
From Jankovic J et al: Bradley and Daroff’s neurology in clinical practice, ed 8, Philadelphia, 2022, Elsevier.
WorkupThe evaluation and management of NF1 patients are summarized in Table E3. Workup of both NF1 and schwannomatosis is largely dictated by clinical symptoms and usually includes MRI evaluation.8
TABLE E3 Evaluation and Management of Neurofibromatosis 1 (NF1) Patients
| At time of diagnosis and annually during childhood and adolescence (unless otherwise indicated) |
Dermatologic examination (especially if a plexiform neurofibroma [PNF] is present)
- Surgical consultation if painful or disfiguring neurofibromas
- PET-CT if rapid growth, increased firmness, or persistent pain within an established PNF or the development of a new neurologic deficit∗
- Ophthalmologic examination with visual assessment (prior to age 8 yr)
- Orbital/brain MRI if signs/symptoms of optic glioma
- Evaluation for scoliosis and tibial bowing
- Referral to orthopedics if scoliosis develops
- Neurologic examination, developmental/behavioral evaluation, and (in young children) measurement of head circumference
- Orbital/brain and/or spinal MRI if neurologic signs/symptoms develop†
- Comprehensive developmental assessment prior to starting school and if problems arise
- Cardiac assessment and measurement of blood pressure
- Referral to cardiology if murmur is detected
- Renal arteriography and 24-hr urine collection for catecholamines and metanephrines if hypertension develops
- Assessment for pubertal development
- Orbital/brain MRI and referral to endocrinology if precocious puberty develops
|
| Minimum annual evaluation for adults with uncomplicated disease |
Dermatologic evaluation if PNF is present
PET-CT for indications described above
Neurologic examination (especially if a PNF is present)
MRI and/or other studies as indicated if neurologic signs/symptoms develop
Measurement of blood pressure
Evaluation as described above if hypertension develops
In women, screening for breast cancer with clinical examination + mammography ± MRI (beginning by age 40 yr) |
| Potentially affected family members |
Dermatologic examination for cutaneous manifestations of NF1
Ophthalmologic examination for Lisch nodules
Consider genetic testing if a mutation has been identified in the proband |
CT, Computed tomography; MRI, magnetic resonance imaging; PET-CT, positron emission tomography-computed tomography.
From Bolognia JL: Dermatology, ed 4, Philadelphia, 2018, Elsevier, pp 985-1003.
Laboratory Tests
- Genetic testing is recommended to distinguish among the various NF conditions.4
- Genetic testing is possible in individuals who desire prenatal diagnosis for NF1. There is no single standard test, and multiple tests are required. Results can tell only if an individual is affected but cannot predict the severity of the disease due to variable expression.
- Pathologic evaluation of tissue samples for definitive tumor typing.9
Imaging Studies
- MRI with and without gadolinium is the imaging study of choice in NF patients. MRI increases detection of optic gliomas, tumors of the spine, acoustic neuromas, and “bright spots” believed to represent hamartomas.
- In NF1, localized MRI is recommended if nonsuperficial plexiform neurofibromas are suspected. XR, CT, and ultrasound may also be indicated based on localized area of interest. The decision to utilize imaging should be directed by clinical symptoms.
- In SWN, baseline MRIs of the brain and spine are recommended. In some cases, whole-body MRI may be indicated for tumor staging and burden.9
Other Tests
- Detailed skin examination; consider utilizing Wood’s lamp in patients with very pale skin for visualizing café-au-lait spots
- Comprehensive neurologic examination
- Complete ophthalmologic examination, including slit lamp
- Audiology examination in NF2-related SWN

Treatment is directed primarily at symptoms and complications.
Nonpharmacologic Therapy
- Counseling addressing prognosis and genetic, psychological, and social issues.
Acute General Rx
- Surgical excision is typically not performed on cutaneous neurofibromas unless cosmetically requested or if suspicion of malignant transformation exists.
- Surgery may be indicated for spinal or cranial neurofibromas, gliomas, or meningiomas.
- Acoustic neuromas can be treated by surgical excision.
- Tumors often cannot be fully resected due to involvement of critical structures, leading to high rates of tumor recurrence.10,11
Chronic Rx
- Adjuvant radiation may be used in advanced disease and can reduce the risk of local recurrence following surgery.
- Carboplatin chemotherapy can treat optic pathway gliomas.
- Kojic acid may reduce hyperpigmentation of Café-au-lait macules.
- Clinical trials involving sirolimus, bevacizumab, and tyrosine kinase inhibitors are in progress.11
- Stereotactic radiosurgery with gamma knife may be an alternative approach to surgery for acoustic neuromas.12
Disposition
- Disposition and prognosis vary according to the severity of symptoms.
- A cure does not exist for NF.
ReferralA multidisciplinary team of consultants is often needed in patients who have NF, including neurosurgeons, otolaryngologists, dermatologists, neurologists, audiologists, speech pathologists, geneticists, neuropsychologists, and pain specialists.
