AUTHOR: Joseph S. Kass, MD, JD, FAAN


Huntington disease (HD) is a trinucleotide repeat autosomal dominant neurodegenerative disorder characterized by involuntary movements manifesting as chorea, psychiatric disturbance, and cognitive decline.
5 to 12 cases/100,000 persons. Most common cause of adult-onset hereditary chorea
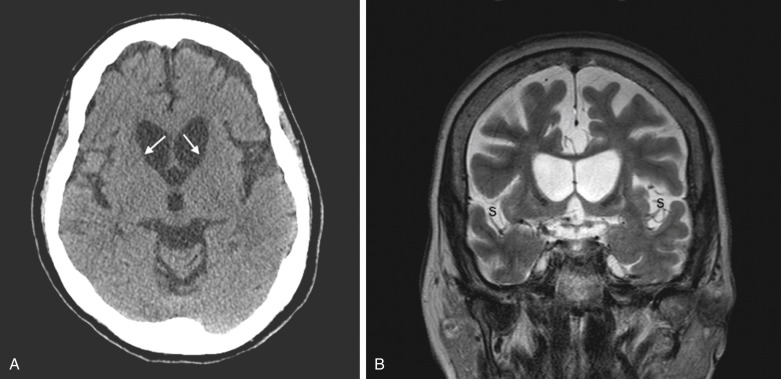
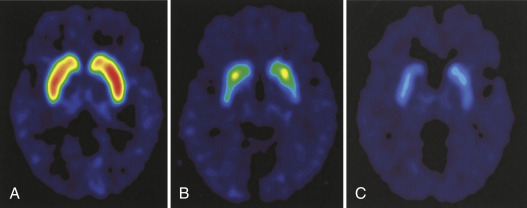
Autosomal dominant due to trinucleotide repeat expansion (CAG) in the huntingtin gene (HTT) on chromosome 4p16.3. Repeat length determines degree of penetrance. See Table E1 for details.
TABLE E1 CAG Repeats and Disease Risk
| CAG Repeat # | Allele Classification | Disease Risk | Risk to Offspring |
|---|---|---|---|
| ≤26 | Normal | No symptoms | None |
| 27-35 | Intermediate | No symptoms | Low unless there is paternal inheritance, which increases risk of anticipation with increased repeat length in offspring that may result in symptomatic disease |
| 36-39 | Disease | At risk for HD | Moderate |
| ≥40 | Disease | HD imminent | High |
HD, Huntington disease.
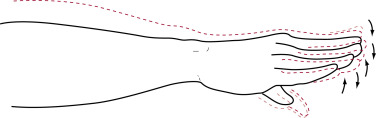
Chorea: hallmark physical manifestation of HD. Chorea refers to involuntary, random, irregular, fragmented, purposeless movements that flow from one body part to the next.
- Body parts affected: limbs, trunk, neck, face, and tongue.
- Velocity varies from fast (mimicking myoclonus) to slow (mimicking dystonia).
- Intensity, frequency, and amplitude may vary from subtle low-amplitude movements in upper face to dramatic large-amplitude movements in the limbs and trunk.
- May initially be mistaken for fidgeting.
- Patients may deny its presence even when obvious to others and may disguise their chorea by incorporating it into a voluntary movement.
- Chorea in HD often involves the upper face and forehead, which is less often involved in other forms of chorea such as tardive dyskinesia.
- Cognitive decline
- Progressive cognitive decline eventually leading to subcortical dementia found in all HD patients.
- Cognitive domains affected: Executive function, planning, working memory, and attention.
- Behavioral affects
- Depression, irritability, anxiety, and impulsivity common
- Some experience obsessive-compulsive symptoms
- Apathy common and worsens over course of disease
- Psychosis is uncommon
- High rates of suicide
- HD prodrome
- May begin up to 10 years before a clinical diagnosis.
- Manifests as subtle motor and nonmotor symptoms not sufficiently significant to yield a clinical diagnosis.
- Cognitive and behavioral symptoms such as depression may precede overt motor symptoms by years and may lead to significant morbidity or even mortality from suicide.
- (Fig. E1). When there is a writhing quality, it is referred to as choreoathetosis. Chorea is present early on and tends to decrease in end stages of disease.
- Dancelike, lurching gait (Fig. E2), often caused by chorea.
- Westphal variant: Typical of juvenile-onset HD and presenting with a parkinsonian syndrome of cognitive dysfunction, bradykinesia, and rigidity.
- Oculomotor abnormalities are common early on and include increased latency of response, hypometric saccades, and insuppressible eye blinking.
- Motor impersistence is common. Patients cannot keep their tongue sticking without moving it in and out of the mouth or cannot hold on to thumb of the examiner for prolonged period of time.
- Psychiatric disorders (can be present early on): Depression is commonly seen, as well as obsessive-compulsive behaviors and aggression associated with impaired impulse control.
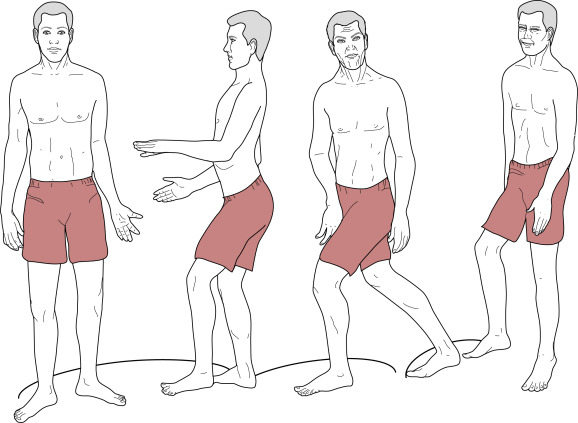
From Kaufman DM et al: Kaufman’s clinical neurology for psychiatrists, ed 8, Philadelphia, 2017, Elsevier.