AUTHORS: Sandeep Soman, MD and Snigdha t. Reddy, MD

Immunoglobulin A nephropathy (IgAN) is a proliferative glomerulonephritis associated with predominant mesangial IgA deposition. IgA deposition can occur in isolation or with IgG, IgM, or both, and often with complement protein C3.
| ||||||||||||||||||||||||
IgA nephropathy (IgAN) is the most common primary glomerulopathy worldwide. The annual incidence of IgAN is about 2.5 per 100,000 person-yr.1,2 There is a very low incidence in Africa relative to the U.S. and a higher incidence in East Asia. In Japan, the incidence is eight times as high as in the U.S.1 Because IgA is the primary enteric antibody, the increased incidence and prevalence in Asia has been postulated to relate to geographic differences in dietary exposure to antigens and genetic factors.
Autopsy studies reveal the prevalence of renal IgA deposits in 1.3% to 16% of cases in the general population.1 Many individuals remain undiagnosed, despite having histologic evidence of IgAN, because they do not have clinical signs of glomerular injury or renal dysfunction. Generally, prevalence rates are expressed as a percentage of cases of primary glomerulonephritis or percentage of total series of renal biopsies. The prevalence in kidney biopsies is 5% in the Middle East, 10% to 35% in Europe, 20% in the U.S., and 32% to 54% in Japan and China. The lower U.S. rates may be biased by a predilection of U.S. nephrologists not to perform kidney biopsy of asymptomatic patients with minimal renal abnormalities.
IgAN is most prevalent in the second and third decades of life with a male:female ratio of 2:1 in the U.S. and 3:1 in Europe. In East Asia, the male:female ratio is closer to 1:1.
Genetic and geographic background predisposes individuals to development of IgAN. The progression of IgAN is highly variable, but approximately 25% of patients develop end-stage renal disease (ESRD) within 10 to 25 yr. The most robust risk factors and predictors of poor renal outcomes at time of diagnosis include older age, hypertension, proteinuria >1 gram per day, and decreased glomerular filtration rate (GFR). Lower hemoglobin and albumin are additional risk factors of poor outcomes. Other remediable risk factors include obesity and smoking. Immunoglobulin levels, specifically IgA antibody, are not required for diagnosis, prognosis, or monitoring of patients.
Pathologic risk factors are based on the Oxford Classification of IgAN (MEST-C) and include increased mesangial cellularity, endocapillary proliferation, segmental glomerulosclerosis, tubular atrophy and interstitial fibrosis, and presence of crescents (see Box E1).3 The score predicts poor outcomes, but isolated endocapillary proliferation does not significantly worsen prognosis. Thrombotic microangiopathy, more commonly encountered in Asians, is associated with worse clinical outcomes.
The primary defect is systemic and caused by an aberrant glycosylation that results in an abnormal antibody, galactose-deficient IgA1 (Gd-IgA1), a heritable trait in diverse racial and ethnic groups. Genetic predisposition is confirmed by elevated serum Gd-IgA1 levels in a quarter of relatives of patients with IgAN. The Gd-IgA1 binds IgG autoantibody and produces renal-inflammatory immune complexes.
Although IgAN is a sporadic disease in more than 90% of cases, recent genome-wide association studies have identified 6% to 8% of cases with at least 15 susceptibility loci, the strongest signal being from the major histocompatibility complex (MHC) region of chromosome 6.2 Certain loci are more prevalent in Asia than in the U.S. and Europe. In addition, Asian Americans have a fourfold higher ESRD incidence than European Americans and sevenfold higher ESRD incidence than African Americans.
- Approximately 75% of children and young adults present with macroscopic hematuria often associated with upper respiratory infection or gastrointestinal illness. This is often referred to as “synpharyngitic hematuria.” Older adults present with microscopic hematuria, proteinuria, and hypertension.
- Loin pain may be associated with macroscopic hematuria. This symptom should not be confused with the much rarer disorder, “loin pain hematuria syndrome.”
- Physical findings are usually unremarkable. Hypertension is present in 20% to 30% of patients with chronic kidney disease, and edema is found in the 5% of patients with nephrotic-range proteinuria.
- IgAN presents as acute kidney injury due to hemoglobin toxicity and intratubular obstruction due to red blood cell casts. It can present as macroscopic hematuria in 5% of patients and as chronic kidney disease in 10% to 20% of patients. Another cause for acute kidney injury can be rapidly progressive glomerulonephritis, frequently associated with the histologic finding of crescents in ≥50% of glomeruli.
- Most cases are idiopathic or primary. IgA vasculitis (formerly Henoch-Schönlein purpura) may represent a more systemic, vasculitic form of IgAN.
- Secondary causes of IgAN (Table E1) include hepatitis B; alcoholic cirrhosis; celiac disease; inflammatory bowel disease (IBD); psoriasis; sarcoidosis; cystic fibrosis; cancer of the lungs, larynx, or pancreas; HIV infection; systemic lupus erythematosus; rheumatoid arthritis; diabetic nephropathy; Sjögren syndrome; and Reiter syndrome. Given the role of IgA in the gut, aside from IBD, infections in the gastrointestinal tract have been associated with IgAN or flares of the disease.
- The four-hit pathogenesis model is depicted in Fig. E1. The first hit from disease heritability is abnormal IgA glycosylation that produces polymeric galactose-deficient antibody, Gd-IgA1. The second hit is when Gd-IgA1 becomes auto-antigenic and auto-antibodies and immune complexes form. The third hit occurs when the immune complexes of Gd-IgA1 and antiglycan antibodies deposit in the mesangium leading to enhanced production of growth factors such as PDGF and IGF-1 with mesangial expansion. The fourth hit is intrarenal complement activation with generation of inflammatory cytokines by activated leukocytes.
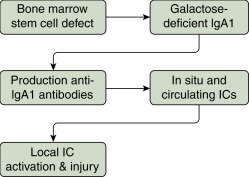
Table E1 Causes of Secondary IgAN
| Group | Disease | ||
|---|---|---|---|
| Gastrointestinal and Liver Disease | Inflammatory bowel disease (Crohn disease, ulcerative colitis), celiac disease, cirrhosis | ||
| Infection | HBV, HCV, HIV, tuberculosis, leprosy | ||
| Autoimmune Diseases | Ankylosing spondylitis, rheumatoid arthritis, Sjogren syndrome | ||
| Malignancy | Lung cancer, renal cell carcinoma, non-Hodgkin and Hodgkin lymphoma, IgA myeloma | ||
| Respiratory tract | Sarcoidosis, bronchiolitis obliterans, pulmonary hemosiderosis, cystic fibrosis, pulmonary fibrosis | ||
| Skin | Dermatitis herpetiformis, psoriasis |
HBV, hepatitis B virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; IgAN, immunoglobulin A nephropathy.
From Pattrapornpisut P et al: IgA nephropathy: core curriculum 2021, Am J Kidney Dis 78(3):429-441, 2021, https://doi.org/10.1053/j.ajkd.2021.01.024.


