AUTHOR: Kapil S. Meleveedu, MD




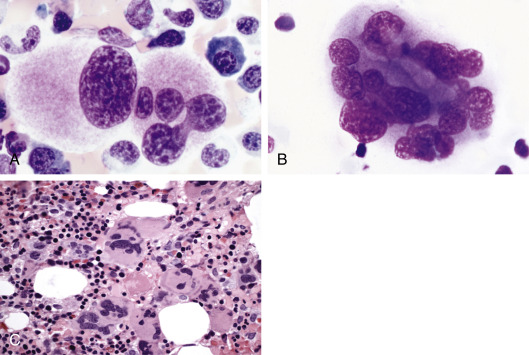
Myelodysplastic syndromes (MDSs) are a group of acquired clonal disorders of hematopoietic stem cells characterized by altered differentiation and proliferation. Presenting features include peripheral blood cytopenias and bone marrow hypercellularity with morphologic abnormalities, which reflects underlying ineffective hematopoiesis with inadequate maturation.
- Earlier classification systems for MDS have included the French-American-British (FAB) classification (1997), the 1997 World Health Organization (WHO) classification, and the 2008 modified WHO classification.
- Currently, the revised 2016 WHO classification is in clinical use (Table 1).
TABLE 1 2016 World Health Organization Classification of the Adult Myelodysplastic Syndromes
| Name | Dysplastic Lineages | Cytopenias∗ | Ring Sideroblasts as % of Marrow Erythroid Elements | BM and PB Blasts | Cytogenetics by Conventional Karyotype Analysis |
|---|---|---|---|---|---|
| MDS with single lineage dysplasia (MDS-SLD) | 1 | 1 or 2 | <15%/<5%† | BM <5%, PB <1%, no Auer rods | Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS with multilineage dysplasia (MDS-MLD) | 2 or 3 | 1-3 | <15%/<5%† | BM <5%, PB <1%, no Auer rods | Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS With Ring Sideroblasts (MDS-RS) | |||||
| MDS-RS with single lineage dysplasia (MDS-RS-SLD) | 1 | 1 or 2 | ≥15%/≥5%† | BM <5%, PB <1%, no Auer rods | Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS-RS with multilineage dysplasia (MDS-RS-MLD) | 2 or 3 | 1-3 | ≥15%/≥5%† | BM <5%, PB <1%, no Auer rods | Any, unless fulfills all criteria for MDS with isolated del(5q) |
| MDS with isolated del(5q) | 1-3 | 1-2 | None or any | BM <5%, PB <1%, no Auer rods | del(5q) alone or with 1 additional abnormality except –7 or del(7q) |
| MDS With Excess Blasts (MDS-EB) | |||||
| MDS-EB-1 | 0-3 | 1-3 | None or any | BM 5%-9% or PB 2%-4%, no Auer rods | Any |
| MDS-EB-2 | 0-3 | 1-3 | None or any | BM 10%-19% or PB 5%-19% or Auer rods | Any |
| MDS, Unclassifiable (MDS-U) | |||||
| With 1% blood blasts | 1-3 | 1-3 | None or any | BM <5%, PB = 1%,‡ no Auer rods | Any |
| With single lineage dysplasia and pancytopenia | 1 | 3 | None or any | BM <5%, PB <1%, no Auer rods | Any |
| Based on defining cytogenetic abnormality | 0 | 1-3 | <15%§ | BM <5%, PB <1%, no Auer rods | MDS-defining abnormality |
| Refractory cytopenia of childhood | 1-3 | 1-3 | None | BM <5%, PB <2% | Any |
∗Cytopenias defined as hemoglobin <10 g/dl; platelet count <100 × 109/L; and absolute neutrophil count <1.8 × 109/L. Rarely, MDS may present with mild anemia or thrombocytopenia above these levels. PB monocytes must be <1 × 109/L.
† If SF3B1 mutation is present.
‡ 1% PB blasts must be recorded on at least 2 separate occasions.
§ Cases with ≥15% ring sideroblasts by definition have significant erythroid dysplasia and are classified as MDS-RS-SLD.
From Arber DA et al: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia, Blood 127(20):2391-2405, 2016.
| ||||||||||||||||
Approximately 4 new cases/100,000 persons per yr. An estimated 30,000 new cases are diagnosed annually in the U.S. The true incidence is likely to be higher because of incomplete case assessment and underreporting of MDS in cancer registries, and it may be close to 75 per 100,000 among persons over the age of 70 years.1
Normal human aging is the most important risk factor for development of MDS due to progressive acquisition of somatic mutations by the hematopoietic stem cells throughout the human life span. Exposure to radiation, chemotherapeutic agents, benzene, or other organic compounds is also associated with myelodysplasia. Table 2 describes predisposing factors and epidemiologic associations of patients with MDS. MDS is extremely rare in pediatric age group but if does arise in patients <18 years of age, work up for inherited or congenital disorders should be sought. With recent advances in genomic sequencing technologies, some individuals with or without cytopenias are found to have somatic clonal mutations which in some cases go on to develop MDS or AML. The understanding regarding these potential premalignant conditions (ICUS, CHIP, CCUS) including factors that determine progression continues to evolve. Up to 40 genes that affect specific functional pathways are mutated in MDS, with 90% of patients having at least one mutation and a median of two to three mutations detected per patient. The most common mutations occur in genes involved in RNA splicing (SF3B1, SRSF2, U2AF1, and ZRSR2), epigenetic modification (TET2, ASXL1, and DNMT3A), regulators of signal transduction (NRAS and JAK2), and transcription factors (RUNX1 and TP53). An increasing number of germline mutations (RUNX1, GATA2, DDX41, etc) are also being identified which are shown to be associated with a familial syndrome with inherited predisposition of developing MDS.
TABLE 2 Predisposing Factors and Epidemiologic Associations of Patients With Myelodysplastic Syndrome
| Heritable | |||
| Constitutional Genetic Disorders | |||
| Trisomy 8 mosaicism Familial monosomy 7 Down syndrome (trisomy 21) Neurofibromatosis 1 Germ cell tumors [embryonal dysgenesis del(12p)] | |||
| Congenital Neutropenia | |||
| Kostmann syndrome Shwachman-Diamond syndrome | |||
| DNA Repair Deficiencies | |||
| Fanconi anemia Ataxia-telangiectasia Bloom syndrome Xeroderma pigmentosum Pharmacogenomic polymorphisms (GSTq1-null) | |||
| Acquired | |||
| Senescence | |||
| Mutagen Exposure | |||
| Alkylator therapy (chlorambucil, cyclophosphamide, melphalan, N-mustards) Topoisomerase II inhibitors (anthracyclines) βEmitters (32p) Autologous stem cell transplantation Environmental/occupational (benzene) Tobacco Aplastic anemia Paroxysmal nocturnal hemoglobinuria |
DNA, Deoxyribonucleic acid.
From Hoffman R et al: Hematology, basic principles and practice, ed 7, Philadelphia, 2018, Churchill Livingstone.