Eveline C. Traeger, MD

DESCRIPTION
- Inherited degenerative storage disorders caused by deficiency of an enzyme that is required for the catabolism of lipids that contain ceramide. The lipids that accumulate in tissues and organs of affected individuals are from the normal turnover of cells and cell components. Differences in properties of the accumulating substances as well as the type of tissue in which a particular lipid component is rapidly turning over account for the diverse clinical manifestation of the disorders.
- There are nine diseases, most of which have variable phenotypes that correlate with the level of residual enzyme activity. Central and/or peripheral nervous system involvement is true of all except type 1 Gaucher and Niemann-Pick type B. The clinical features described in this chapter are for the most common phenotype with neurologic manifestations. Phenotypes that may be characterized by delayed onset during adolescence or adulthood and usually associated with variable neurologic and systemic manifestations are described in more extensive reviews.
EPIDEMIOLOGY
Incidence/Prevalence
- Tay Sachs (GM2-Gangliosidosis, Type I): Incidence is highest among people of Ashkenazi Jewish descent with a carrier rate of 1 in 27 people. Incidence is also increased in the Pennsylvania Dutch group, French-Canadians in Quebec, and Cajun community of Louisiana. In the general population, 1 in 250 people are carriers.
- Sandhoff (GM2-Gangliosidosis, Type II): Incidence of 1/384,000 live births. Creole population in Argentina, Métis Indians in Saskatchewan, and individuals of Lebanese heritage have a higher incidence.
- GM1-gangliosidosis: Estimated incidence is 1:100,000-200,000. A high incidence of 1/3,700 is reported in the population of Malta.
- Fabry: Prevalence estimated between 1/40,000 and 1/117,000.
- Niemann-Pick type A: Panethnic but with an increased incidence in Ashkenazi Jews of 1/40,000.
- Gaucher: Panethnic. Type I is most commonly diagnosed in Ashkenazi Jews. Type 3 is more common in the Norrbottnian region of Sweden.
- Farber Lipogranulomatosis: Very rare. Incidence is not known.
- Krabbe: Panethnic with an incidence estimated at 1/100,000 births. Incidence of 6/1,000 births in some Arab communities in Israel. Incidence also increased in the Scandinavian countries, reported as
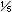 0,000.
0,000. - Metachromatic Leukodystrophy: Panethnic with an incidence estimated at 1/40,000 to 160,000 births. A particularly high incidence of 1/75 in the Habbanite Jewish community in Israel. Incidence of 1/2,500 in the western portion of the Navajo Nation and 1/8,000 among Arab groups in Israel.
Sex
Because of X-linked inheritance, patients with Fabry are male. Female heterozygotes may manifest symptoms of the disease but symptoms are less severe and of later onset.
RISK FACTORS
Genetics
The sphingolipidoses are inherited in an autosomal-recessive manner except for Fabry, which is X-linked. Carrier identification and prenatal testing are available.
PATHOPHYSIOLOGY
Deficiency of a lysosomal hydrolase required to degrade glycosphingolipids, an essential component of cell membranes. Lysosomal accumulation of the enzyme's substrate results in physiologic and morphologic alterations of specific tissues and organs with clinical manifestations that may include neurodegeneration, organomegaly, skeletal abnormalities, bone marrow dysfunction and pulmonary infiltration.

HISTORY
- Tay-Sachs and Sandhoff: Onset at 3 to 5 months with exaggerated startle response to sound and decreased visual attentiveness. By 6 to 10 months of age, there is progressive weakness and loss of previously attained milestones. Thereafter, progression is rapid. A cherry-red spot is present in almost all patients. Seizures usually develop by the end of the first year. Macrocephaly from reactive cerebral gliosis is common. Organomegaly is a feature of Sandhoff disease, but not Tay Sachs disease.
- GM1-gangliosidosis: Onset of developmental arrest before 6 months of age followed by progressive CNS deterioration. Of patients, 50% have a cherry-red spot. Hepatosplenomegaly is almost always present. Skeletal dysplasia seen. Patients become vegetative with generalized spasticity, contractures, and generalized seizures.
- Fabry disease: Onset in preteen and adolescent boys. Early signs of characteristic corneal and lenticular opacities, small punctuate reddish-blue angiokeratoma on the umbilicus, flank, thighs, penis and scrotum, pain in the extremities (acroparesthesia), and hypohidrosis. Progressive CNS damage from prothrombotic and occlusive abnormalities, and large vessel ectasias with transient ischemic attacks, vascular thromboses, seizures, hemorrhagic or ischemic strokes. Progressive cardiac and renal disease.
- Niemann-Pick type A: Onset prior to 6 months of age with psychomotor retardation. A cherry-red spot is present in 50% of patients. Progressive spasticity, rigidity, and vegetative state. Hepatosplenomegaly.
- Gaucher disease type 2: Onset from infancy to 6 months of age with progressive CNS damage including marked mental retardation, seizures, hypertonicity with hyperactive reflexes, cranial nerve involvement with strabismus, facial weakness, and dysphagia. Hepatosplenomegaly.
- Farber lipogranulomatosis: Onset from infancy to 4 months of age. Swollen, painful joints with subcutaneous nodules over affected joints and pressure points. Progressive aphonia, swallowing, and feeding difficulties due to laryngeal involvement. Lower motor neuron involvement, which manifests as hypotonia and muscular atrophy. Psychomotor development variable from severe involvement to normal intelligence. Cherry-red spot macula.
- Krabbe: Onset 3 to 6 months of age with psychomotor delay, tonic seizures, progressive motor impairment with hypertonicity. Deafness and blindness are common. Peripheral neuropathy detected. CSF protein increased. Clinical symptoms restricted to nervous system.
- Metachromatic leukodystrophy: Late infantile form with onset at age 1 to 2 years, with progressive ataxia, hypotonia, and diminished deep tendon reflexes. Progressive optic atrophy and spastic quadriparesis. Slowing of conduction velocities of peripheral nerve. CSF protein increased.
PHYSICAL EXAM
- Progressive neurodegeneration with loss of previously attained milestones.
- Macrocephaly in GM2-Gangliosidosis
- Organomegaly in Sandhoff, GM1 gangliosidosis, Niemann-Pick type A, Gaucher type 2.
- A cherry-red spot macula on ophthalmologic exam in GM2 and GM1-Gangliosidosis, Niemann-Pick type A and Farber disease.
- Skeletal abnormalities in GM1 gangliosidosis.
- Angiokeratoma in Fabry disease.
Imaging
Initial Approach
- Neuroimaging studies may reveal nonspecific changes such as atrophy.
- Skeletal radiographs for patients with GM1 gangliosidosis may reveal anterior beaking of vertebrae, enlargement of sella turcica, and thickening of calvaria.
Diagnostic Procedures/Other
- Diagnosis is made by enzymatic assay of the specific enzyme in leukocytes, or skin fibroblasts.
- Tay Sachs: Hexosaminidase A deficiency
- Sandhoff: Hexosaminidase A and B deficiency
- GM1: β-galactosidase deficiency
- Fabry: α-galactosidase A (ceramide trihexosidase) deficiency
- Niemann-Pick type A: Sphingomyelinase deficiency
- Gaucher: Glucocerebrosidase deficiency
- Farber: Ceramidase deficiency
- Krabbe: Galactocerebrosidase deficiency
- Metachromatic Leukodystrophy: Arylsulfatase A deficiency
DIFFERENTIAL DIAGNOSIS
The sphingolipidoses must be differentiated from other inherited neurodegenerative diseases.

MEDICATION
First Line
- Enzyme replacement therapy (ERT) with recombinant glucocerebrosidase for patients with Gaucher type 2 and type 3 disease as a palliative measure to treat severe visceral involvement. Treatment does not alter the neurologic progression.
- ERT with recombinant alpha-galactosidase A for patients with Fabry disease.
- There are no specific treatments for the other disorders.
COMPLEMENTARY AND ALTERNATIVE THERAPIES
- Symptomatic Treatment
- Carbamazepine or phenytoin, occasionally in combination with amitriptyline, is used to treat the painful neuropathy in patients with Fabry disease.
- Kidney transplant and long-term hemodialysis in patients with Fabry disease with renal failure.
- Bone marrow transplant for treatment of Gaucher type 3 may decrease visceral storage.
- Adjunctive Treatment
- The indication for physical therapy should be assessed on an individual basis.
IN-PATIENT CONSIDERATIONS
Admission Criteria
Patients are usually admitted for evaluation and treatment of the neurologic and systemic complications of their disorder.

PATIENT MONITORING
Patient follow-up is guided by the predicted course and potential complications of the disease.
PATIENT EDUCATION
- United Leukodystrophy Foundation, 2304 Highland Dr., Sycamore, IL 60178. Phone: 800-728-5483.
- National Tay-Sachs and Allied Diseases Association, 2001 Beacon St., Ste. 204, Brighton, MA 02135. Phone: 800-90-NTSAD.
- National Gaucher Foundation, 11140 Rockville Pike, Ste. 350, Rockville, MD 20852-3106. Phone: 800-925-8885.
PROGNOSIS
- Tay Sachs and Sandhoff: Vegetative state rapidly ensues with death by 2 to 4 years of age.
- GM1: Death ensues a few years after onset of the disease.
- Fabry: Death usually occurs from renal failure, cardiovascular involvement, or cerebrovascular disease. Average age at death is 41 years.
- Niemann-Pick type A: Death occurs by 2 to 3 years of age.
- Gaucher type 2: Death in infancy.
- Farber: Death in late infancy or early childhood.
- Krabbe: Death in infancy or early childhood.
- Metachromatic leukodystrophy: Death 1 to 7 years after onset.
