= brain cell death leading to coagulation necrosis
Cause: large vessel occlusion of ICA / MCA / PCA (50%) ← emboli from atherosclerotic stenosis, small-vessel lacunes (25%), cardiac cause (15%), blood disorder (5%), non-arteriosclerotic (5%), venous thrombosis (1%)
- 33% of TIAs will lead to infarction!
Pathophysiology:
distal microstasis occurs within 2 minutes after occlusion of cerebral artery; regional cerebral blood flow is acutely decreased in area of infarction + remains depressed for several days at center of infarct; arterial circulation time may be prolonged in entire hemisphere; rapid development of vasodilatation due to hypoxia, hypercapnia, tissue acidosis; delayed filling + emptying of arterial channels in area of infarction (= arteriolar-capillary block) well into venous phase; by end of 1st week regional blood flow commonly increases to rates even above those required for metabolic needs (= hyperemic phase = luxury perfusion)
- stroke
Mimics: intracerebral hemorrhage, subdural hematoma, cerebritis, hemiplegic / hemisensory migraine, tumor, arteriovenous malformation
Detection rate by CT:
80% for cortex + mantle, 55% for basal ganglia, 54% for posterior fossa
- Positive correlation between degree of clinical deficit and CT sensitivity
- CT sensitivity: on day of ictus 48%
- 1–2 days later 59%
- 7–10 days later 66%
- 10–11 days later 74%
Location: cerebrum÷cerebellum = 19÷1
- supratentorial
- cerebral mantle (70%) in territory of MCA (50%), PCA (10%), watershed between MCA + ACA (7%), ACA (4%)
- basal ganglia + internal capsule (20%)
- infratentorial (10%) upper cerebellum (5%), lower cerebellum (3%), pons + medulla (2%)
Hyperacute Ischemic Infarction
Time period:<12 hr
NECT (100% specific):
Sensitivity: 57% (with standard 40/20 HU window width & level; increased to 71% with 8/32 HU)
- normal (in 10–60%)
- identifies hemorrhage
- Hemorrhage contraindicates thrombolytic therapy!
- subtle decrease in attenuation within affected brain area due to cytotoxic edema:
- “disappearing basal ganglia” sign = obscuration and loss of gray-white matter differentiation in basal ganglia:
- lentiform nucleus becomes isodense to internal and external capsule within 2 hours after onset of stroke
- “insular ribbon” sign = loss of gray-white matter differentiation of insular cortex (in 50–80% of MCA occlusions)
- “disappearing basal ganglia” sign = obscuration and loss of gray-white matter differentiation in basal ganglia:
- “hyperdense vessel” sign (HIGHLY SPECIFIC):
- “hyperdense middle cerebral artery” sign = acute intraluminal thrombus of 80 HU (← extrusion of serum from thrombus) versus 40 HU of flowing blood in M1 segment; transient phenomenon
Incidence: 17–35–50% of acute MCA occlusions
Associated with: poor clinical outcome
DDx: high hematocrit, polycythemia, calcification of vessel wall (usually bilateral), arterial dolichoectasia - “MCA dot” sign = punctate focus of hyperattenuation in sylvian fissure (in M2/M3 segment of MCA) ← thromboembolus (38% sensitive, 100% specific)
- calcified intraluminal embolus (rare)
- hypoattenuating fat embolus (rare)
- “hyperdense middle cerebral artery” sign = acute intraluminal thrombus of 80 HU (← extrusion of serum from thrombus) versus 40 HU of flowing blood in M1 segment; transient phenomenon
- mass effect from brain swelling:
- hemispheric cortical sulcal effacement / compression
- narrowing of sylvian fissure (in MCA infarct)
CECT:
- visualization of thrombus within intracranial arteries
- assessment of carotid + vertebral arteries
Perfusion CT:
= monitoring first pass of a small iodinated contrast agent bolus through cerebral circulation with continuous cine imaging for 45 seconds of same slab of tissue
- Motion artifacts in ROI invalidate the study!
- Time-attenuation curves for arterial ROI in unaffected ACA / MCA + venous ROI in superior sagittal sinus / torcular Herophili and in each parenchymal pixel
- Color-coded perfusion maps for
- mean transit time (MTT)
= mathematical deconvolution on time-attenuation curve of each pixel with respect to arterial curve- Most sensitive to disruption of cerebral perfusion
- MTT correlates directly with perfusion pressure
Increase in: ischemia (infarct core and penumbra), asymptomatic vessel stenosis, vasospasm - cerebral blood volume (CBV)
= dividing areas under curve in parenchymal pixel by area under curve in arterial pixel- CBV higher in highly vascularized basal ganglia and cortical surface than in white matter
- increased CBV in penumbra → functioning autoregulation → dilatation of vessels
- decreased CBV in infarct core → loss of autoregulation → blood vessels no longer dilate
- cerebral blood flow (CBF = CBV÷MTT)
- Most important parameter!
Normal:>50 mL / 100 g brain tissue
Ischemia threshold: 10–15 mL / 100 g
- mean transit time (MTT)
- Summary map of percentage of mismatch
= CBV area ÷ CBF area- ischemic area (= prolonged MTT) divided into:
- irreversibly infarcted nonsalvageable tissue (“infarct core”)
- markedly decreased CBF + markedly decreased CBV
- surrounded by stunned (nonfunctioning potentially salvageable) cells that receive collateral blood supply (“penumbra”)
- decreased CBF + normal / mildly increased CBV ← autoregulatory mechanisms
- irreversibly infarcted nonsalvageable tissue (“infarct core”)
- ischemic area (= prolonged MTT) divided into:
Perfusion CT Analysis of Hyperacute Ischemic Stroke
|
MR (routinely positive by 4–6 hours post ictus):
- parenchymal changes:
- bright signal (← less signal loss) on DWI with ↓ ADC
Pathophysiology of homeostasis of tissue water:- excess intracellular water (= cytotoxic edema) + ↓ rate of water molecular diffusion (= restriction of normal brownian motion of water molecules)
Sensitivity & specificity: 88–100% & 86–100%
Rule of thumb: low signal intensity on ADC map means the stroke is <1 week old! - hyperintense signal on T2WI + FLAIR involving cortical gray matter
In most ischemic strokes FLAIR images are positive 6–12 hours after onset of symptoms! - loss of gray-white matter differentiation on T2WI
- subtle parenchymal swelling with sulcal effacement ← cytotoxic edema can be seen by 2 hours post ictus (best on T1WI)
- abnormal blooming on T2*WI identifies hemorrhage
The diagnosis of ischemic stroke is unlikely if parenchymal enhancement persists >8–12 weeks.
- bright signal (← less signal loss) on DWI with ↓ ADC
- vessel signs:
- loss of normal intravascular flow voids on T2WI
- intravascular low SI on T2* + high SI on FLAIR (similar to “hyperdense MCA” sign)
- stasis of contrast material within affected arteries ← stasis of flow distal to thrombus
- ischemic penumbra = combination of perfusion + diffusion-weighted images allows identification of areas at risk for infarction
DWI & ADC Map in Acute Stroke
|
NUC:
- Newer imaging agents (eg, 99mTc-HMPAO) may be positive within minutes of the event, while CT and MR are normal
- hemispheric hypoperfusion throughout all phases
- defect corresponding to nonperfused vascular territory
- “flip-flop” sign in radionuclide angiogram (15%)
= decreased uptake during arterial + capillary phase followed by increased uptake during venous phase - “luxury perfusion syndrome” (14%) = increased perfusion
Rx: recombinant tissue plasminogen activator (tPA) if symptom onset <6 hours ago
One-third rule: early ischemic changes on NECT in >⅓ of MCA territory (in acute ischemic stroke within 6 hours of symptom onset) excludes patient for IV and intraarterial thrombolytic therapy → NO benefit with higher risk of symptomatic hemorrhage
Histo: cortical cytotoxic edema (= accumulation of intracellular water due to cell membrane damage) followed by extracellular white matter vasogenic edema
- bright lesion on DWI very conspicuous within 0–6 hours after onset of symptoms + up to 14 days after ictus (diffusion coefficient is a measure of proton mobility in tissue)
- false positive DWI:
diffusion coefficient of infarcts is influenced by T2 properties + b-value of gradient strength (T2 shine-through) - 5% false negative DWI
- false positive DWI:
- hypointense signal on ADC map (negates T2 shine-through effect) within 24 hours
(ADC map shows pure diffusion characteristics without T2 effect, but has low lesion conspicuity)- a low coefficient (acute infarct) gives a hyperintense signal
- a high coefficient (CSF) gives a hypointense signal
Early Acute Ischemic Infarction
Time period: 12–24 hours
NECT:
- low-density lesion (30–60% invisible)
- loss of differentiation between cortical gray matter and subjacent white matter:
- blurring of the clarity of internal capsule
- “insular ribbon” sign = hypodense extreme capsule no longer distinguishable from insular cortex
- subtle sulcal effacement (8%)
CECT:
- no iodine accumulation in affected cortical region
- meningeal gyriform enhancement
MR:
- subtle narrowing of sulci
- blurring of gray-white matter junction on T2- and proton-density images
- increase in thickness of cortex (= gyral swelling)
- subtle low signal intensity on T1WI, high signal intensity on T2WI (masking of gyral infarcts on heavily T2WI ← sulcal CSF intensity)
- 20–30% false negative T2WI during first 24 hours
MRA:
- absence of flow for infarcts >2 cm in diameter
Late Acute Ischemic Infarction
Time period: 1–3–7 days
NECT:
- hypodense wedge-shaped lesion with base at cortex in a vascular distribution (in 70%) ← vasogenic + cytotoxic edema
- mass effect (23–75%): sulcal effacement, transtentorial herniation, displaced subarachnoid cisterns + ventricles
- “bland infarct” may be transformed into hemorrhagic infarct after 2–4 days ← leakage of blood from ischemically damaged capillary endothelium following lysis of intraluminal clot + arterial reperfusion
CECT:
- ⇓meningeal and ⇓ intravascular contrast enhancement
- ⇑parenchymal enhancement
MR:
- ↑SI on DWI during 1st week after onset of symptoms
- “intravascular enhancement” sign (77%)
= Gd-pentetate enhancement of cortical arterial vessels in area of brain injury after 1–3 days ← slow arterial blood flow provided by collateral circulation via leptomeningeal anastomoses - “meningeal enhancement” sign (33%)
= Gd-pentetate enhancement of meninges adjacent to infarct after 2–6 days ← meningeal inflammation
Angio:
- narrowed / occluded vessels supplying infarcted area
- delayed filling + emptying of involved vessels
- early draining vein
- luxury perfusion of infarcted area (rare) = loss of small vessel autoregulation ← local increase in pH
Time period: 7–30 days = paradoxical phase with resolution of edema + onset of coagulation necrosis
NECT:
- “fogging phenomenon” = less apparent low-density area
- decrease of mass effect + ex vacuo dilatation of ventricles (in 57%)
- ± transient calcification (especially in children)
CECT:
- gyral blush + ring enhancement for 2–8 weeks (in 65–80% within first 4 weeks) ← breakdown of blood-brain barrier + luxury perfusion
- NO enhancement in 20% of patients
MR:
Histo: vasogenic edema (= increased extracellular water) ← disruption of blood-brain barrier
- ↑SI on DWI ← T2 shine-through from infarcted tissue
- hypointense on T1WI, hyperintense on T2WI
- intravascular + meningeal enhancement signs resolve toward end of 1st week
- gyriform parenchymal Gd-pentetate enhancement
- Gyriform parenchymal enhancement permits differentiation of subacute from chronic infarction!
- infarction flip-flops from hyperintense lesion to iso- / hypointense lesion on ADC maps 5–10 days after ictus
Time period: months to years (>30 days)
Histo: demyelination + gliosis complete
◊Focal brain atrophy after 8 weeks!
- cerebral atrophy + encephalomalacia + gliosis (HALLMARKS)
- possible calcification (especially in children)
NECT:
- cystic foci of CSF density (= encephalomalacia) in distribution of vascular territory
MR:
- patchy region with increased intensity on T2WI
- gliosis (hyperintense on T2WI) often surrounding encephalomalacic region
- wallerian degeneration (= antegrade degeneration of axons ← neuronal injury) of corticospinal tracts in the wake of old large infarcts that involve the motor cortex
Hemorrhagic transformation is rare during first 12 hours after stroke onset and usually occurs within 24–48 hours. It is almost always present 4–5 days after stroke.
Etiology: lysis of embolus / opening of collaterals / restoration of normal blood pressure following hypotension / hypertension / anticoagulation → extravasation in reperfused ischemic brain
Incidence: 6% of clinically diagnosed brain infarcts; 20% of autopsied brain infarcts
Path: petechial hemorrhages in various degrees of coalescence
Location: corticomedullary junction
CT:
- hyperdensity (56–76 HU) appearing within a previously imaged hypodense area of acute ischemic infarction = hemorrhagic transformation (in 50–72%)
False negative: hematoma isoattenuating if hematocrit <20%
MR:
- hypointense area on T2WI within edema marking gyri = deoxyhemoglobin of acute hemorrhage
- hyperintense area on T1WI = methemoglobin of subacute hematoma
- Early parenchymal enhancement within 6 hours of stroke has a higher risk for clinically significant hemorrhagic transformation.
= occlusion of small penetrating arteries at base of brain (lenticulostriate / thalamoperforating arteries) = lacunar infarct (= infarct <1 cm in size)
Cause:
- Embolism
- Hypoperfusion
- Carbon monoxide poisoning
- Drowning
- Vasculopathy (hypertension, microvasculopathy, aging)
- dense homogeneous enhancement outlining caudate nucleus, putamen, globus pallidus, thalamus
- dense round nodular enhancement / peripheral ring enhancement
= ischemic changes affecting deep layers of the cortex
◊Layers 3, 5, 6 are very sensitive to oxygen deprivation
MR:
- acute stage
- linear cortical hyperintensity on T1WI
- contrast enhancement
- white matter edema on T2WI
- chronic stage
- thin hypointense cortex
- hyperintense white matter
- enlargement of CSF spaces
[lacuna, Latin = hole]
= small deep infarcts in the distal distribution of penetrating vessels (lenticulostriate, thalamoperforating, pontine perforating arteries, recurrent artery of Heubner)
Cause: occlusion of small penetrating end arteries arising from MCA, PCA, basilar, ACA and vertebral arteries at base of brain due to fibrinoid degeneration
Age: usually >55 years; M÷F = 1÷1
Predisposed: hypertension, diabetes
Incidence: 15–20% of all strokes
Path: lacune = small hole of encephalomalacia traversed by cobweblike fibrous strands; if multiple = état lacunaire (lacunar state)
Histo: “microatheroma” = hyalinization + arteriolar sclerosis → thickening of vessel wall + luminal narrowing
- pure motor / pure sensory stroke
- ataxic hemiparesis, vascular dementia
Location: upper two-thirds of putamen >caudate >thalamus >pons >internal capsule
- small discrete foci of hypodensity of 3–15 mm in size (most <1 cm in diameter)
MR:
- acute lacunar infarction (between 12 hours and 7 days):
- small high-signal–intensity region on T2WI + FLAIR
- hypointense area on T1WI
- high signal intensity on DWI + corresponding low signal intensity on ADC map
- chronic lacunar infarction:
- high signal intensity on T2WI
- low signal intensity on T1WI
- hypointense center + hyperintense rim (= gliosis) on FLAIR
- normal signal intensity on DWI
- may enhance in late acute / early subacute stage (up to 8 weeks)
- unilateral pontine infarcts are sharply marginated at midline
DDx: enlarged Virchow-Robin spaces, neurocysticercosis
- hypodense small lesions located peripherally near / within cortex without enhancement
- lesions detected in only 14%, contralateral lesion present in 14% (CT of marginal value)
Watershed / Border Zone Infarct
= infarct localized to border zone between 2 adjacent nonanastomosing vascular beds of major cerebral arteries
Cause: global hypoperfusion ← poor cardiac output / cervical carotid artery occlusion or severe stenosis / noncompetent circle of Willis / microembolism
Isolated cortical border zone infarcts may be embolic in nature and are less frequently associated with hemodynamic compromise.
Pathophysiology:
- repeated episodes of hypotension with severe arterial stenosis / occlusion → lower perfusion pressure → autoregulatory vasodilation → increased susceptibility to ischemia → infarction (esp. for external border zone infarcts)
- Stage I hemodynamic impairment:
- ↑cerebral blood volume + ↑ mean transit time
- Stage II hemodynamic impairment:
- ↓blood flow + ↑ oxygen extraction (measured with PET) = misery perfusion= chronic failure of cerebral autoregulation
Classification of Border Zone Infarcts
External (cortical) Infarct Frontal cortex (between ACA + MCA) Occipital cortex (between MCA + PCA) Paramedian white matter (between ACA + MCA) Internal (subcortical) Infarct Between lenticulostriate + MCA Between lenticulostriate + ACA Between Heubner artery + ACA Between anterior choroidal artery + MCA Between anterior choroidal artery + PCA - Stage I hemodynamic impairment:
- microemboli from heart / atherosclerotic plaques in major arteries → preferential propagation to cortical border zones secondary to lower perfusion pressure → limited ability to wash out emboli (esp for internal border zone infarcts)
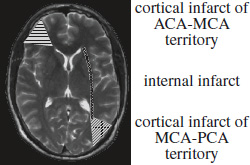
Location:
- cortical (external) watershed
Cause: (a) Unilateral posterior external border zone infarcts ← emboli from heart / CCA
(b) Bilateral infarcts ← vascular stenosis- wedge-shaped / ovoid cortical “territorial” infarct
- location of cortical border zones may vary ← development of leptomeningeal collaterals
- subcortical (internal) watershed
Cause: arterial stenosis / occlusion / hemodynamic compromise.- multiple 3-mm infarcts in rosarylike pattern arranged in linear fashion parallel to lateral ventricle in centrum semiovale / corona radiata
Small internal border zone infarcts typically represent the “tip of the iceberg” of decreased perfusion reserve and may be predictive of impending stroke.
- 6% of cerebral infarcts are hemorrhagic (red infarct)
- completed stroke, TIA, RIND
- amaurosis fugax = transient monocular blindness
- weakness / numbness in an extremity, aphasia, dizziness
- diplopia, dysarthria (vertebrobasilar ischemia)