Image
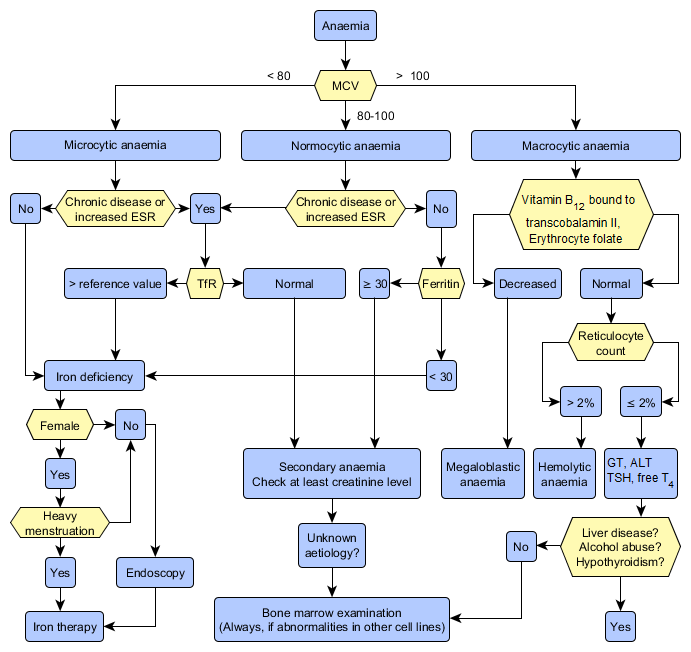
Examination of Anaemia

- Women 117-155 g/l, men 134-167 g/l
- Microcytic anaemia: iron deficiency anaemia, anaemia of chronic disease (ACD, secondary anaemia), thalassaemia
- Normocytic anaemia: anaemia of chronic disease, haemolytic anaemia, acute bleeding, aplastic anaemia, bone marrow infiltration
- Macrocytic anaemia: vitamin B12 or folate deficiency, marked reticulocytosis, blood loss (> 2 days), liver disease, heavy alcohol consumption, myelodysplasia and other haematological malignancy, hypothyroidism
Iron deficiency anaemia
- Iron deficiency anaemia is a symptom, and the underlying diagnosis should be determined.
- Iron deficiency is always abnormal in women over reproductive age and in all men.
- If the patient has microcytic anaemia and secondary anaemia appears to be unlikely, iron deficiency anaemia is very likely.
- Additional investigations
- Plasma ferritin< 30 µg/l is a sign of iron deficiency. A normal or increased value may be due to an inflammatory reaction (ferritin is an acute-phase reactant) and does not exclude iron deficiency in a patient with elevated CRP or ESR.
- Plasma soluble transferrin receptor (TfR) assay is used to measure iron deficiency associated with a chronic disease. A value exceeding the upper limit of reference range is strongly suggestive of iron deficiency.
- The cause of iron deficiency should be determined with gastrointestinal endoscopy in all men and in those women whose anaemia is not due to menstrual bleeding.
Anaemia of chronic disease (ACD)
- Chronic infection and other inflammatory conditions, malignancy
- Organ specific aetiology: most commonly chronic renal failure, liver disease
- First-line investigations: full blood count, ESR, CRP
- Bone marrow examination is rarely needed in the investigation of anaemia. Bone marrow examination is indicated in unclear cases if the cause of the anaemia is not revealed through blood tests and the patient has no systemic disease that causes chronic anaemia.
- Suspect megaloblastic anaemia if the patient presents with macrocytic anaemia (increased erythrocyte MCV).
- Establish the aetiology of anaemia (usually vitamin B12 or folate deficiency).
- Vitamin B12 deficiency: atrophic gastritis (pernicious anaemia, Helicobacter), gastrectomy, disease affecting distal small bowel, tapeworm infestation
- Folate deficiency: dietary deficiency (alcoholics), increased requirement (pregnancy, prematurity, haemolysis, cancer), malabsorption (coeliac disease), increased loss (some skin and liver diseases, dialysis), several medicines
Haemolytic anaemia
- Haemolytic anaemias are very common worldwide (sickle cell anaemia, thalassaemias, abnormal haemoglobins, autoimmune haemolytic anaemia [AIHA] etc.).
- Diagnostic workout should include a reticulocyte count, in addition to full blood count. A normal erythrocyte reticulocyte count usually excludes the possibility of haemolysis.