Treatment of Hypertension JNC 7

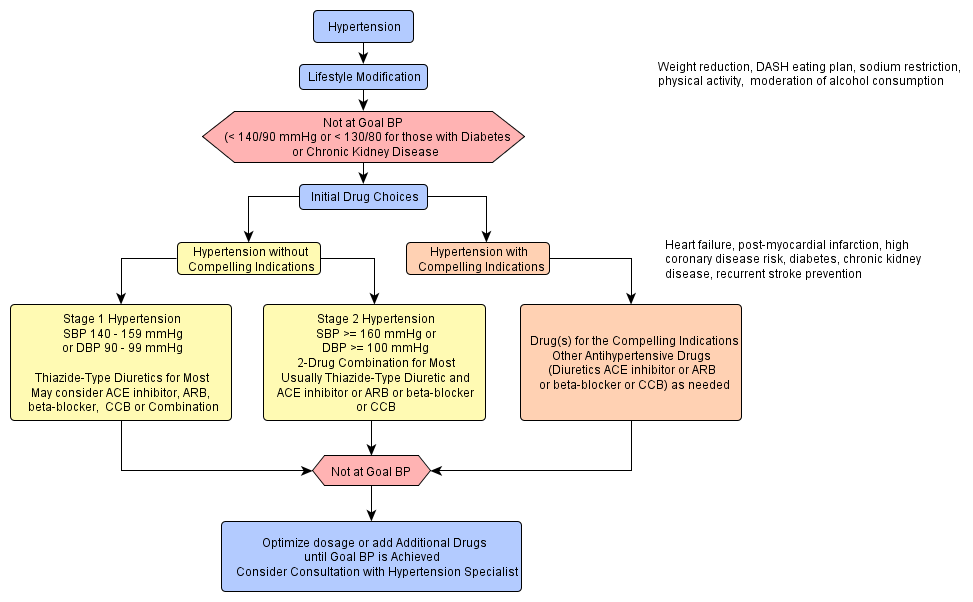
"The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure" http://www.nhlbi.nih.gov/guidelines/hypertension/express.pdf provides a new guideline for hypertension prevention and management. The following are the key messages(1) In persons older than 50 years, systolic blood pressure (BP) of more than 140 mm Hg is a much more important cardiovascular disease (CVD) risk factor than diastolic BP;
(2) The risk of CVD, beginning at 115/75 mm Hg, doubles with each increment of 20/10 mm Hg; individuals who are normotensive at 55 years of age have a 90% lifetime risk for developing hypertension;
(3) Individuals with a systolic BP of 120 to 139 mm Hg or a diastolic BP of 80 to 89 mm Hg should be considered as prehypertensive and require health-promoting lifestyle modifications to prevent CVD;
(4) Thiazide-type diuretics should be used in drug treatment for most patients with uncomplicated hypertension, either alone or combined with drugs from other classes. Certain high-risk conditions are compelling indications for the initial use of other antihypertensive drug classes (angiotensin-converting enzyme inhibitors, angiotensin-receptor blockers, beta-blockers, calcium channel blockers);
(5) Most patients with hypertension will require 2 or more antihypertensive medications to achieve goal BP (<140/90 mm Hg, or <130/80 mm Hg for patients with diabetes or chronic kidney disease);
(6) If BP is more than 20/10 mm Hg above goal BP, consideration should be given to initiating therapy with 2 agents, 1 of which usually should be a thiazide-type diuretic; and
(7) The most effective therapy prescribed by the most careful clinician will control hypertension only if patients are motivated. Motivation improves when patients have positive experiences with and trust in the clinician. Empathy builds trust and is a potent motivator. Finally, in presenting these guidelines, the committee recognizes that the responsible physician's judgment remains paramount.