The Hgb molecule consists of four polypeptide globin chains and four heme components containing iron and the red pigment porphyrin. Hemoglobin formation is genetically determined, and the types of globin chains normally formed are termed alpha (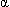 ), beta (
), beta (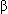 ), gamma (
), gamma (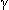 ), and delta (
), and delta (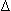 ). Combinations of these chains form various types of Hgb. Disorders of synthesis and production of globin chains result in the formation of abnormal Hgb.
). Combinations of these chains form various types of Hgb. Disorders of synthesis and production of globin chains result in the formation of abnormal Hgb.
Hemoglobin electrophoresis is a technique for identifying the types of Hgb present and for determining the percentage of each type. Exposed to an electrical current, the several types of Hgb migrate toward the positive pole at different rates. The patterns created are compared with standard patterns.
At birth, most RBCs contain fetal hemoglobin (Hgb F), which is made up of two chains and two chains. Within a few months, through sequential suppression and activation of individual genes, Hgb F largely disappears and is replaced by adult hemoglobin (Hgb A). Hgb A, composed of two chains and two chains, makes up more than 95 percent of Hgb in adults. A minor type of Hgb, Hgb A2, consisting of two chains and two chains, also is found in small amounts (2 to 3 percent) in adults. Traces of Hgb F persist throughout life (Fig. 1—4).29
More than 150 genetic abnormalities in the Hgb molecule have been identified. These are termed thalassemias and hemoglobinopathies. Thalassemias are genetic disorders in globin chain synthesis that result in decreased production rates of - or -globin chains. Hemoglobinopathies refer to disorders involving an abnormal amino acid sequence in the globin chains.
In -thalassemia, for example, production of chains and Hgb A is decreased. The oversupply of chains results in the formation of hemoglobin H (Hgb H), which consists of four chains (Fig. 1—5). Complete absence of a chain production (homozygous thalassemia A) is incompatible with life and generally results in stillbirth during the second trimester of pregnancy. The cord blood of such fetuses shows high levels of hemoglobin Barts, a type of Hgb that evolves from unpaired chains. Hemoglobin Barts itself has such a high affinity for oxygen that it releases none to the tissues.
In -thalassemia minor, a decrease is seen in -chain production and, therefore, a reduction in the amount of Hgb A formed. In -thalassemia major, all -chain production is lost and no Hgb A is formed. The chains are then used to form Hgb F and Hgb A2.
Among the most common Hgb abnormalities are the sickle cell disorders, which exhibit a double gene defect that results in the production of hemoglobin S (Hgb S). In Hgb S, the amino acid valine is substituted for glutamine at a critical position on the globin chain, which causes the chains to "lock" when deoxygenated, deforming the erythrocyte into the sickled shape. Repeated sickling damages RBC membranes and shortens the cells' life spans. The abnormally shaped cells pass more sluggishly through the circulation, leading to impaired tissue oxygenation.
The gene for Hgb S is most prevalent in black populations and may be present as either sickle cell trait (having one recessive gene for Hgb S) or sickle cell disease (having both recessive genes for Hgb S). The Sickledex test, a screening test for sickle cell disorders, detects sickled erythrocytes under conditions of oxygen deprivation. Hemoglobin electrophoresis is necessary, however, to differentiate sickle cell trait (20 to 40 percent Hgb S) from sickle cell disease (70 percent Hgb S).
Many other types of abnormal Hgb are caused by defects in globin chain synthesis. Hemoglobin C (Hgb C), for example, has an abnormal amino acid substitution on the chain and can lead to a form of mild hemolytic anemia. Other examples of abnormal Hgb resulting from rearrangement or substitution of the amino acids on the globin chains include hemoglobin E (Hgb E), hemoglobin Lepore (-chain abnormalities), and hemoglobin Constant Spring (-chain abnormality).30
Other disorders involving Hgb pertain to the oxygen-combining ability of the heme portion of the molecule. Examples of types of Hgb associated with such disorders are methemoglobin (Hgb M), sulfhemoglobin, and carboxyhemoglobin. Hgb M is formed when the iron contained in the heme portion of the Hgb molecule is oxidized to a ferric instead of a ferrous form, thus impairing its oxygen-combining ability. Methemoglobinemia may be hereditary or acquired. The acquired form may be caused by excessive radiation or by the toxic effects of chemicals and drugs (e.g., nitrates, phenacetin, lidocaine). Note that Hgb F is more easily converted to Hgb M than is Hgb A.
Sulfhemoglobin is a pigment that results from Hgb combining with inorganic sulfides. It occurs in those who take sulfonamides or acetanilid.
Carboxyhemoglobin results when Hgb is exposed to carbon monoxide. Although this type of Hgb is most commonly seen in individuals with excessive exposure to automobile exhaust fumes, it may also occur in heavy smokers.31 Tests other than Hgb electrophoresis are used to determine the presence of Hgb M and carboxyhemoglobin.