Chromatin Structure and Epigenetic Marks.
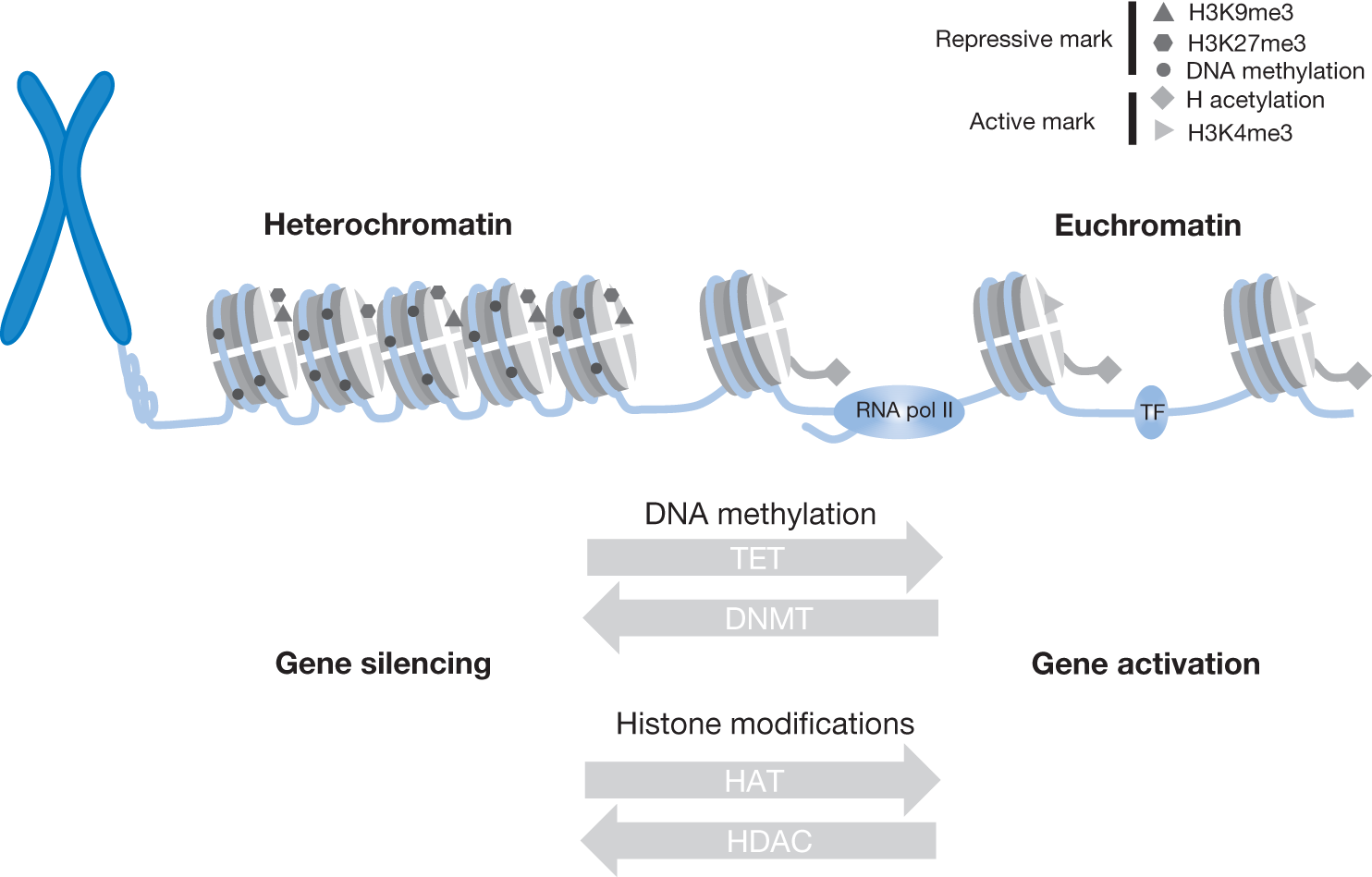
Chromatin remodeling and bidirectional transition from transcriptionally active euchromatin to compacted heterochromatin is orchestrated by multiple epigenetic modifications at DNA and nucleosome levels. DNA methylation at CpG sites is generally associated with genes silencing, and the methylation pattern is heritable during cell division. This process is catalyzed by 1 of 3 DNA methyltransferases identified in mammals. DNMT1 is considered a main maintenance methyltransferase and is responsible for preserving the methylation pattern after cell division. DNMT3A and DNMT3B are so-called de novo methyltransferases that establish a DNA methylation pattern during development. DNA methylation is a physiologically reversible process. TET genes are members of enzymes called dioxygenases. TET1 and TET2 catalyze the conversion of 5-methylcytosine to 5-hydroxymethylcytosine that constitutes the initial step of DNA demethylation. Histone modifications represent the second level of epigenetic control of the overall transcriptional program. Over 200 histone modifications have been described. The most common and best understood are histone methylation and acetylation. The latter process is usually associated with relaxation of chromatin that facilitates access of transcription factors (TF) and RNA polymerase complex to DNA. Histone acetylation is catalyzed by histone acetyltransferases (HAT). Conversely, histone deacetylation catalyzed by histone deacetylases (HDAC) marks chromatin compaction (heterochromatin) and subsequent inhibition of transcription. Activation and inhibition of gene expression is also dependent on methylation of histone tails either on lysine (K) or on arginine (R) residues. The methylation pattern commonly associated with active transcription include H3K4, H3K48, and H3K79. Transcription repression is achieved by methylation of H3K9 and H3K27.