AUTHORS: Brett Patrick, MD and Mark F. Brady, MD, MPH, MMSc and Fred F. Ferri, MD




Pheochromocytomas are catecholamine-producing tumors that originate from the chromaffin cells of the adrenergic system. While they generally secrete both norepinephrine and epinephrine, norepinephrine is usually the predominant amine.
| ||||||||||||||||
- Incidence: 0.05% of population; peak incidence in 30s and 40s.
- Approximately 25% of patients with apparently sporadic pheochromocytoma may be carriers of mutations.
- Approximately 25% of pheochromocytomas are familial and associated with genetic disorders (Table 1). Pheochromocytoma is a feature of two disorders with an autosomal dominant pattern of inheritance:
- Pheochromocytomas occur in 5% of patients with neurofibromatosis type 1.
TABLE 1 Autosomal Dominant Syndromes Associated With Pheochromocytoma and Paraganglioma
| Syndrome | Gene | Gene Locus | Protein Product | Protein Function | Gene Mechanism | Typical Tumor Location |
|---|---|---|---|---|---|---|
| SDHD (familial paraganglioma type 1)∗ | SDHD | 11q23 | SDH D subunit | ATP production | Tumor suppressor | Skull base and neck; occasionally adrenal medulla, mediastinum, abdomen, pelvis |
| Familial paraganglioma type 2∗ | SDHAF2 | 11q13.1 | Flavination cofactor | ATP production | Tumor suppressor | Skull base and neck; occasionally abdomen and pelvis |
| SDHC (familial paraganglioma type 3) | SDHC | 1q21 | SDH C subunit | ATP production | Tumor suppressor | Skull base and neck |
| SDHB (familial paraganglioma type 4) | SDHB | 1p36.1-35 | SDH B subunit | ATP production | Tumor suppressor | Abdomen, pelvis and mediastinum; rarely adrenal medulla, skull base, and neck |
| MEN-1 | MEN-1 | 11q13 | Menin | Transcription regulation | Tumor suppressor | Adrenal medulla |
| MEN-2A and MEN-2B | RET | 10q11.2 | RET | Tyrosine kinase receptor | Protooncogene | Adrenal medulla, bilaterally |
| Neurofibromatosis type 1 | NF1 | 17q11.2 | Neurofibromin | GTP hydrolysis | Tumor suppressor | Adrenal-periadrenal |
| von Hippel-Lindau disease | VHL | 3p25-26 | VHL | Transcription elongation suppression | Tumor suppressor | Adrenal medulla, bilaterally; occasionally paraganglioma |
| Familial pheochromocytoma | FP/TMEM127 | 2q11 | Transmembrane protein | Regulation of the mTORC1 signaling complex | Tumor suppressor | Adrenal medulla |
ATP,Adenosine triphosphate; GTP, guanosine triphosphate; MEN, multiple endocrine neoplasia; mTORC1, mammalian target of rapamycin complex 1; RET, “rearranged during transfection” proto-oncogene; SDH, succinate dehydrogenase; VHL, von Hippel-Lindau disease.
∗Associated with maternal imprinting.
From Melmed S: Williams textbook of endocrinology, ed 12, Philadelphia, 2011, Saunders.
- Hypertension: Can be sustained (55%) or paroxysmal (45%).
- Headache (80%): Usually paroxysmal in nature and described as “pounding” and severe.
- Palpitations (70%): Can be present with or without tachycardia.
- Hyperhidrosis (60%): Most evident during paroxysmal attacks of hypertension.
- Physical examination may be entirely normal if done in a symptom-free interval; during a paroxysm the patient may demonstrate marked increase in both systolic and diastolic pressure, profuse sweating, visual disturbances (caused by hypertensive retinopathy), dilated pupils (from catecholamine excess), paresthesias in the lower extremities (caused by severe vasoconstriction), tremor, and tachycardia.
- Orthostatic hypotension is common among patients with pheochromocytoma due to reduction of blood volume and desensitization of adrenergic receptors by the chronic excess of catecholamines.
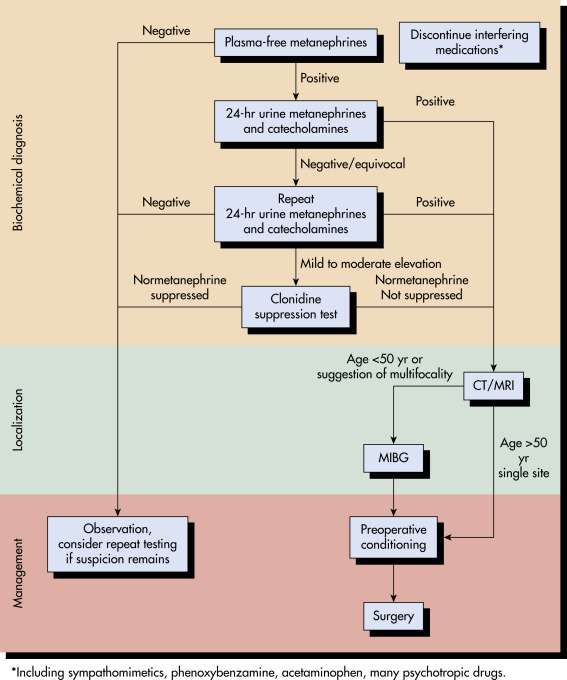
- Box E1 summarizes features suggestive of pheochromocytoma.
BOX E1 Features Suggestive of Pheochromocytoma
Hypertension, Persistent or Paroxysmal
|
From Zipes DP: Braunwald’s heart disease: a textbook of cardiovascular medicine, ed 11, Philadelphia, 2019, Elsevier.
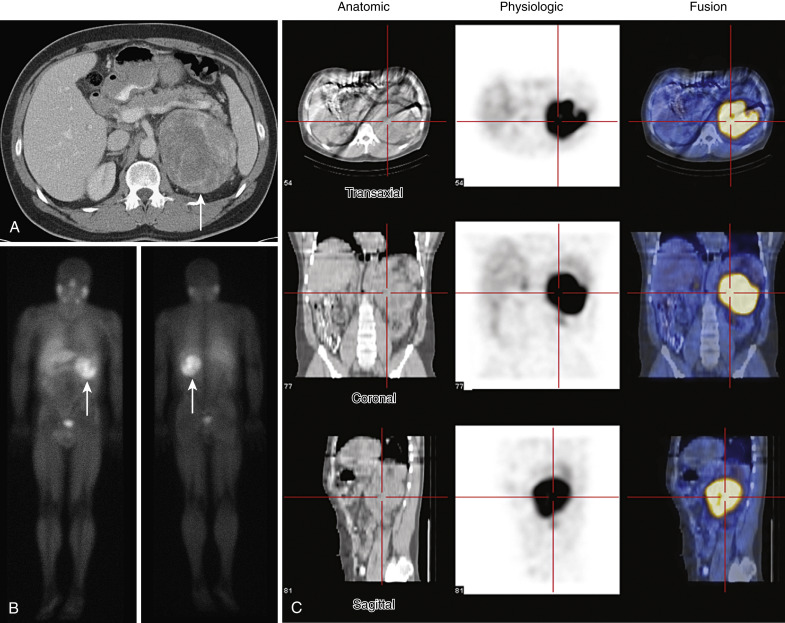
- Catecholamine-producing tumors that are usually located in the adrenal medulla.
- Specific mutations of the RET protooncogene cause familial predisposition to pheochromocytoma in MEN-2.
- Mutations in the von Hippel-Lindau tumor suppressor gene (VHL gene) cause familial disposition to pheochromocytoma in von Hippel-Lindau disease.
- Recently identified genes for succinate dehydrogenase subunit D (SDHD) and succinate dehydrogenase subunit B (SDHB) predispose carriers to pheochromocytoma and globus tumors.