AUTHORS: Hannah Sweeney, MD, and Sarah Hall, MD


Definition
Acromegaly is a chronic disease that develops due to hypersecretion of growth hormone (GH), which stimulates secretion of insulin-like growth factor (IGF-1). It is a progressive, debilitating disease with an insidious onset and potentially lethal outcome.
| ||||||||
Epidemiology & Demographics
Physical Findings & Clinical Presentation
- Coarse features resulting from growth of soft tissue
- Coarse, oily skin
- Macrocephaly
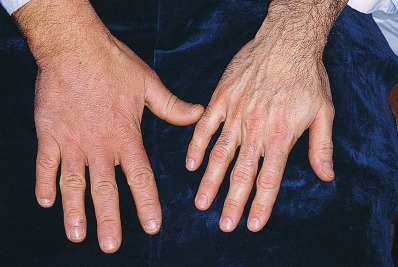
- Hands and feet that are spadelike (Fig. E1), fleshy, and moist
- Prognathism, which can give an underbite
- Excessive sweating
- Menstrual dysfunction
- History of increasing hat, glove, and/or shoe size
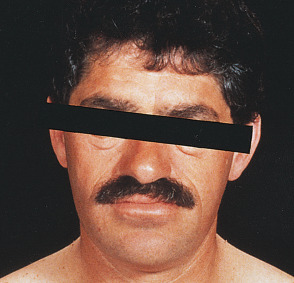
- Enlargement of nose, brow, and jaw (Fig. E2)
- Muscle weakness and decreased exercise capacity
- Headache, often severe
- Visual field defects
- Arthralgias and severe osteoarthritis
- Carpal tunnel syndrome
- Diabetes mellitus
- Hypertension
- Severe obstructive sleep apnea due to soft tissue swelling and macroglossia
- Cardiac abnormalities (cardiomyopathy, left ventricular hypertrophy, valvular heart disease, diastolic failure, arrhythmias)
- Mild to moderate obesity
- Vertebral fractures
- Benign tumors (uterine myoma, skin tags, prostatic hypertrophy, colon polyps)