AUTHOR: Harinder P. Singh, MD


Cystic fibrosis (CF) is an autosomal recessive disorder characterized by dysfunction of exocrine glands that involves multiple organ systems but chiefly results in chronic respiratory infections and pancreatic enzyme insufficiency.
| ||||||||||||||||||||||||
- CF is the most common life-shortening genetic disease in Caucasians (one case per approximately 3200 births in Caucasians) and the second most common life-shortening childhood-onset inherited disorder in the U.S., behind sickle cell disease.
- Worldwide it affects 80,000 children and adults.
- Median age at diagnosis is 5.3 mo. Median survival is now >40 yr because of advancements in treatment.
- Carrier screening is associated with a decrease in incidence of CF.
- Meconium ileus
- Failure to thrive in children
- Increased anterior/posterior chest diameter
- Basilar crackles and hyperresonance to percussion
- Digital clubbing
- Chronic cough
- Abdominal distention
- Greasy, foul-smelling feces
- Clinical manifestations of CF are summarized in Box E1. Atypical presentations are summarized in Table E1
- The frequency of selected GI manifestations in CF is summarized in Table E2
- See Box E1
TABLE E2 Frequency of Selected GI Manifestations in CF∗
| Organ | Manifestation | Frequency in All Patients (%) | Frequency in Adults (%) |
|---|---|---|---|
| Pancreas | Total achylia | 85-90 | 85-90∗ |
| Abnormal glucose tolerance | 20-30 | 20-30 | |
| Partial or normal function | 10-15 | 10-15 | |
| Pancreatitis | 1-2 (all CF) 22 (PS-CF) | 2-3 | |
| Diabetes mellitus | 4-7 | 4-7 | |
| Intestine | Meconium ileus | 10-25 | |
| Rectal prolapse | 1-2 | ||
| Distal intestinal obstruction syndrome | 3 | 18 | |
| Intussusception | 1 | 1-2 | |
| Liver | Fatty liver | 7 | 20-60 |
| Focal biliary cirrhosis | 2-3 | 11-70 | |
| Portal hypertension | 2-3 | 28 | |
| Biliary tract | Gallbladder abnormal, nonfunctional, or small | 25 | 5-20 |
| Gallstones | 8 | 10-25 | |
| Bile duct strictures | 1-20 | 1-20 | |
| Esophagus | GERD | Unknown | 80 |
CF, Cystic fibrosis; GERD, gastroesophageal reflux disease; GI, gastrointestinal; PS-CF, CF with pancreatic sufficiency.
∗Frequency may depend on the genotype.
From Feldman M et al: Sleisenger and Fortran’s gastrointestinal and liver disease, ed 10, Philadelphia, 2016, Elsevier.
TABLE E1 Atypical Presentations of Cystic Fibrosis∗
| Respiratory | |||
| Gastrointestinal |
| ||
| Genitourinary | |||
| Other |
∗Bold type signifies possible presentation in adolescents or adults with cystic fibrosis.
From Broaddus VC et al: Murray & Nadel’s textbook of respiratory medicine, ed 7, Philadelphia, 2022, Elsevier.
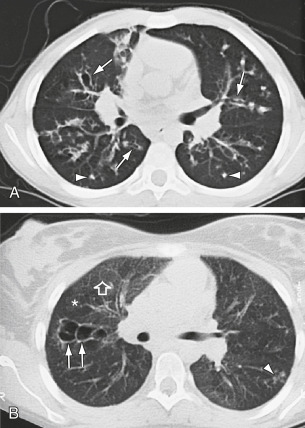
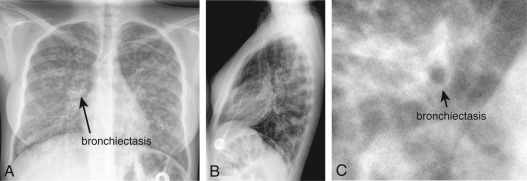
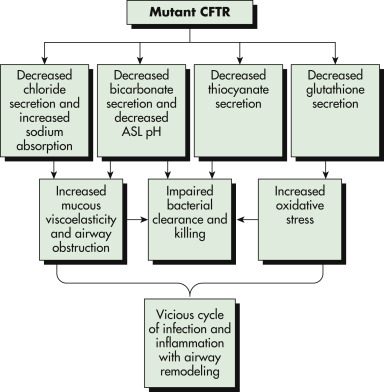
CF is an autosomal-recessive disease caused by mutations to the gene encoding for the CF transmembrane conductance regulator (CFTR) protein (Fig. E1) on chromosome 7; gene mutations organized into six classes (Table E3) have been associated with CF. About half of patients in the U.S. with CF are homozygous for the Phe508del mutation in CFTR, and >90% have at least one Phe508del allele. These mutations result in abnormalities in chloride transport and thus water flux across the surface of epithelial cells (Figs. E2 and E3); the resulting abnormally viscous secretions cause obstruction of glands and ducts in various organs (Fig. E4) and subsequent damage to exocrine tissue (recurrent pneumonia, atelectasis, bronchiectasis, diabetes mellitus, biliary cirrhosis, cholelithiasis, intestinal obstruction, increased risk of GI malignancies). The airway of CF patients shows an exuberant hyperinflammatory response to infection leading to progressive lung damage.
TABLE E3 Classes of Cystic Fibrosis Transmembrane Conductance Regulator Mutations
| Class | Effect on CFTR | Functional CFTR | Presence of CFTR on Cell Membrane |
|---|---|---|---|
| I | Defective protein production due to premature termination of CFTR messenger RNA | No | No |
| II | Impaired protein processing due to misfolding (e.g., ΔF508 deletion) | No | No, CFTR is degraded in the cytoplasm |
| III | Defective regulation with reduced channel opening time (e.g., G551D mutation) | No | Yes |
| IV | Impaired function causing reduced chloride transport | Yes, but reduced in function | Yes |
| V | Reduced synthesis of normally functioning CFTR | Yes, but reduced in number | Reduced in number |
| VI | Impaired membrane insertion or stability | Yes, but reduced in number | Reduced in number |
CFTR, Cystic fibrosis transmembrane conductance regulator; RNA, ribonucleic acid.
From Weinberger SE: Principles of pulmonary medicine, ed 7, Philadelphia, 2019, Elsevier.
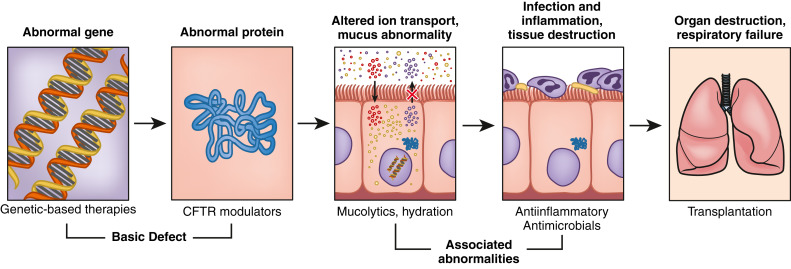
Figure E4 Spectrum of cystic fibrosis disorders.
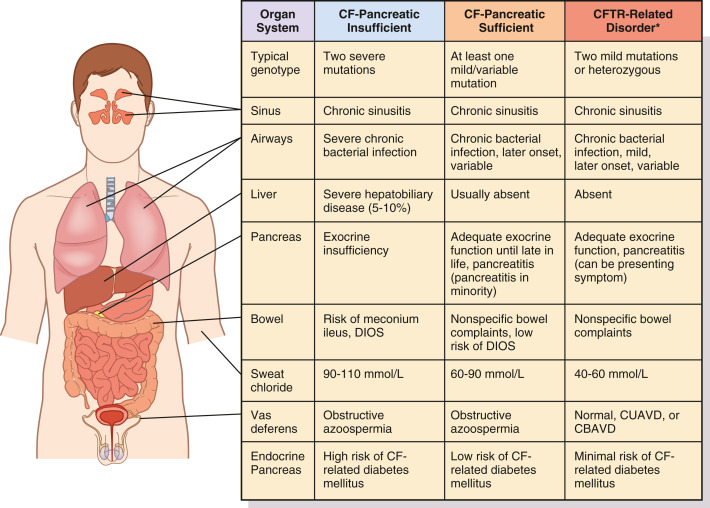
A comparison between findings in severe cystic fibrosis (CF) and milder forms of the disease is shown. Although manifestations are variable, severity in each organ system is generally consistent with degree of cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction conferred by genotype. CBAVD, Congenital bilateral absence of the vas deferens; CUAVD, congenital unilateral absence of the vas deferens; DIOS, distal intestinal obstruction syndrome. ∗Refers to CFTR-related metabolic syndrome or CFTR-related disorders.
From Broaddus VC et al: Murray & Nadel’s textbook of respiratory medicine, ed 7, Philadelphia, 2022, Elsevier.
Figure E3 Schematic of the mucociliary transport defect in cystic fibrosis (CF).
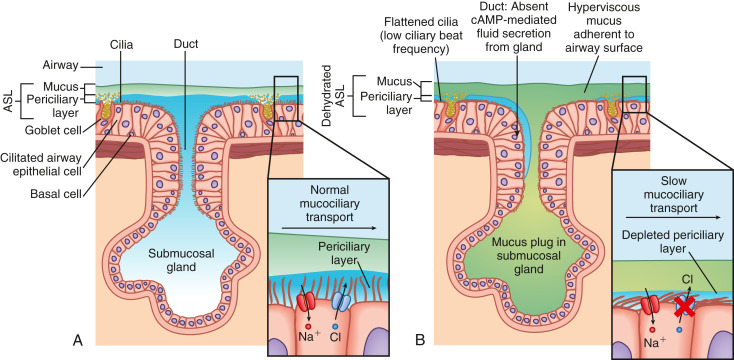
(A) In the healthy state, adequate airway surface homeostasis ensures effective transport of mucus extruding from airway surface goblet cells and the submucosal glands. The airway surface liquid (ASL) is maintained by fluid secretion via the cystic fibrosis transmembrane conductance regulator (CFTR) and fluid absorption via the epithelial sodium channel (ENaC) (inset at right) (CFTR, surface receptor in blue; ENaC, surface receptor in red). (B) In CF, the ASL is depleted through the absence of CFTR-mediated fluid secretion, accompanied by tonic fluid absorption via the ENaC. CFTR-dependent liquid dehydration decreases the depth of the ASL, including the periciliary layer, causing abnormal clearance of mucus from the epithelial cell surface in the airways. cAMP, Cyclic monophosphate.
From Broaddus VC et al: Murray & Nadel’s textbook of respiratory medicine, ed 7, Philadelphia, 2022, Elsevier.
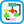
CFTR conducts several anions including chloride, bicarbonate, thiocyanate, and glutathione. The loss of CFTR function affects critical airway epithelial functions: (1) It increases the risk for dehydration of airway surface liquid (ASL) with loss of chloride efflux and associated increased sodium channel activity. (2) The loss of secreted bicarbonate and/or acidic pH of the ASL increases mucous viscoelasticity, resulting in failure of mucociliary transport. (3) Acidic pH in the ASL impairs normal innate immune clearance of bacteria. (4) Loss of thiocyanate impairs lactoperoxidase bacterial killing. (5) Loss of glutathione secretion depletes the antioxidant capacity of the airway, resulting in increased inflammation, increased mucous secretion, and increased mucous viscoelasticity. These factors lead to a vicious cycle of infection and inflammation that is progressive. ASL, Airway surface liquid; CFTR, cystic fibrosis transmembrane conductance regulator.
From Kleiman RM: Nelson textbook of pediatrics, ed 21, Philadelphia, 2020, Elsevier.
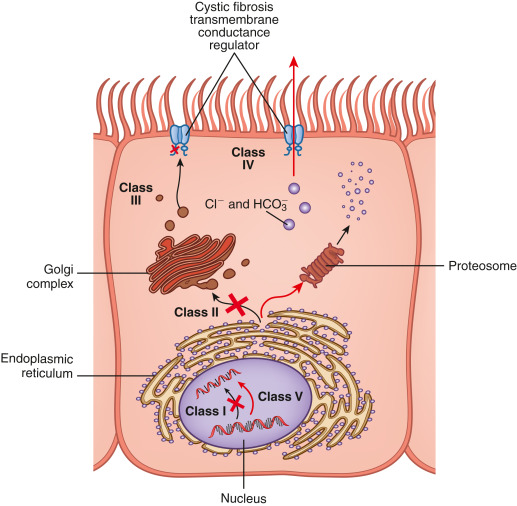
Classes of defects in the CFTR gene product include the absence of synthesis (class I); defective protein maturation and premature degradation (class II); disordered regulation, such as diminished adenosine triphosphate binding and hydrolysis (class III); defective chloride conductance or channel gating (class IV); and a reduced number of CFTR transcripts due to a promoter or splicing abnormality (class V). Another class of defect has reduced protein stability at the cell surface (class VI, not shown).
From Broaddus VC et al: Murray & Nadel’s textbook of respiratory medicine, ed 7, Philadelphia, 2022, Elsevier.