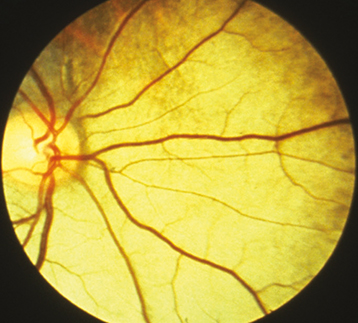
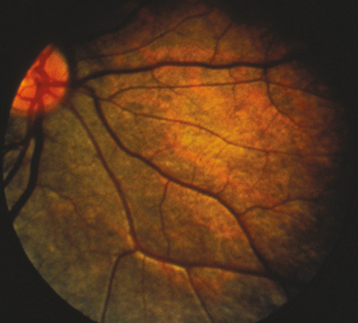
RETINITIS PIGMENTOSA
Decreased night vision (nyctalopia) and loss of peripheral vision in rod–cone dystrophy (RCD) and decreased vision in the light (hemeralopia) with cone–rod dystrophy (CRD) forms of RP. Decrease in central vision can occur early or late in the disease process depending on whether rod or cone involvement is predominant. The same is true for color vision.
(See Figure 11.28.1.)
Clumps of pigment dispersed throughout the peripheral retina in a perivascular pattern, often assuming a “bone spicule” arrangement (though bone spicules may be absent), areas of RPE depigmentation or atrophy, arteriolar narrowing; later, waxy optic disc pallor. Patients often have mild vitreous cells. Progressive visual field loss, usually a ring scotoma, which progresses to a small central field. ERG usually moderately to markedly reduced.
Focal or sectoral pigment clumping, CME, ERM, posterior subcapsular cataract, optic disc drusen.
Autosomal recessive (most common): Diminished vision (severe) and night blindness occur early in life. Asking about family history, including consanguinity, is important.
Autosomal dominant (least severe): More gradual onset of RP, typically in adult life, variable penetrance, late onset of cataract. Visual loss less severe. Can have sectoral RP especially in the inferior retina.
X-linked recessive (rarest and most severe): Onset similar to autosomal recessive. Female carriers often have salt-and-pepper fundus. Visual loss is severe.
Some evidence supports that diets high in vitamin A (15,000 IU/d) and lutein (12 mg/d) may help slow midperipheral visual field loss in nonsmokers with RP, but the literature remains inconclusive. Recent evidence suggests that high-dose vitamin A is not helpful. Both epiretinal and subretinal microchip implants have been used with some success to improve vision in patients with very advanced RP but are presently not commercially available. Clinical trials are ongoing with a variety of designs to determine the safety and efficacy of retinal implant technology. In addition, gene therapy has been approved for RPE65 form of RP and other gene therapy trials are ongoing for RCD and CRD.
SYSTEMIC DISEASES ASSOCIATED WITH HEREDITARY RETINAL DEGENERATION
It is important to ask all patients with suspected inherited retinal dystrophies about possible systemic associations. The most common are genitourinary abnormalities, polydactyly, supernumerary digits, and hearing loss. Other important organ systems that can be associated with inherited retinal disorders are cardiac and central nervous system.
Refsum Disease (Phytanoyl-CoA Hydroxylase Deficiency)
Autosomal recessive RP (often without bone spicules) with increased serum phytanic acid level. May have cerebellar ataxia, peripheral neuropathy, deafness, dry skin, anosmia, liver disease, and cardiac abnormalities. Treat with low-phytanic acid, low-phytol diet (minimize the amount of milk products, animal fats, and green leafy vegetables). Check serum phytanic acid levels every 6 months.
Hereditary Abetalipoproteinemia (Bassen–Kornzweig Syndrome)
Autosomal recessive RP (usually without bone spicules) with fat intolerance, diarrhea, crenated erythrocytes (acanthocytes), ataxia, progressive restriction of ocular motility, and other neurologic symptoms as a result of deficiency in lipoproteins and malabsorption of the fat-soluble vitamins (A, D, E, and K). Diagnosis based on serum apolipoprotein-B deficiency.
Group of autosomal recessive retinal dystrophies that represent the most common genetic cause of congenital blindness in children. Fundus appearance is variable but typically shows a pigmentary retinopathy. Moderate-to-severe vision loss identified at or within a few months of birth, infantile nystagmus, poor and/or paradoxical pupillary response, photophobia, oculodigital sign (eye poking), and markedly reduced or flat ERG. Associated with keratoconus. Voretigene neparvovec (Luxturna) is the first FDA-approved gene therapy for any inherited retinal degeneration. Treatment for the RPE65 variant of Leber congenital amaurosis (LCA) involves subretinal injection of an AAV2 viral vector carrying RPE65 DNA. Severe early-onset CRD dystrophy (SECORD) and early-onset CRD dystrophy (ECORD) are milder versions of LCA that have later onset than LCA.
Multiple subtypes exist, all autosomal recessive. Associated with congenital sensorineural hearing loss which is usually stable throughout adult life. Genes involved code a protein complex present in inner ear hair cells and retinal photoreceptor cells. Molecular testing for certain subtypes is available. Type 1 is associated with congenital hearing loss requiring cochlear implants. Type 2 usually (Ush2a) is more common and has less severe hearing loss.
Mainly autosomal recessive group of different diseases with similar findings including pigmentary retinopathy, hypogonadism, obesity, polydactyly, mental retardation, and others. FDA has approved setmelanotide to treat the obesity in Bardet–Biedl syndrome. Lawrence–Moon syndrome is a related but separate entity associated with spastic paraplegia, but without the polydactyly and obesity.
Salt-and-pepper pigmentary degeneration of the retina with normal arterioles. Progressive limitation of ocular movement without diplopia, ptosis, short stature, endocrinopathies, and/or cardiac conduction defects. Ocular signs usually appear before age 20 years. Mitochondrial inheritance and usually a new mutation with large deletions in the mitochondrial genome. Refer the patient to a cardiologist. Patients may need a pacemaker. See 10.12, Chronic Progressive External Ophthalmoplegia.
Spielmeyer–Vogt–Batten–Mayou syndrome: Associated with seizures, dementia, and ataxia.
Alström and Cockayne syndromes: Associated with hearing loss.
Zellweger syndrome: Associated with hypotonia, hypertelorism, and hepatomegaly.
Maternally inherited diabetes and deafness (mitochondrially inherited): Looks like pentosan toxicity, see below.
Bietti crystalline dystrophy: Autosomal recessive condition characterized by crystals of unknown composition in the peripheral corneal stroma and in the retina at different layers. Can cause choroidal atrophy, decreased night vision, decreased visual acuity, and a flat ERG.
Neuronal ceroid lipofuscinosis type 2 can look like Stargardt but starts at a slightly younger age; no treatment is presently available for this systemic disease.
Enhanced S cone syndrome: Do not make rod pigment or medium and long cone pigment so only blue cones are present. ERG is characteristic with the combined rod and cone response equal to the single photopic cone response.
Thioridazine: Pigment clumps between the posterior pole and the equator, areas of retinal depigmentation, retinal edema, visual field abnormalities (central scotoma and general constriction), depressed or extinguished ERG. Symptoms and signs may occur within weeks of starting phenothiazine therapy, particularly if very large doses (≥2,000 mg/d) are taken. Usually, more than 800 mg/d chronically needed for toxicity. Discontinue if toxicity develops. Follow every 6 months.
Chlorpromazine: Abnormal pigmentation of the eyelids, cornea, conjunctiva (especially within the palpebral fissure), and anterior lens capsule; anterior and posterior subcapsular cataract; rarely, a pigmentary retinopathy with visual field and ERG changes similar to that described for thioridazine. Usually, 1,200 to 2,400 mg/d for longer than 12 months needed for toxicity. Discontinue if toxicity develops. Follow every 6 months.
Pentosan polysulfate toxicity: Oral medication used for the treatment of interstitial cystitis. Multifocal, vitelliform-like deposits at level of RPE (parafoveally), paracentral RPE atrophy. Early cases exhibit parafoveal, punctate, orange, hyperreflective lesions on multicolor OCT and near-infrared reflectance (NIR). Fundus autofluorescence shows speckled hyper- and hypoautofluorescence, with advanced cases showing larger areas of atrophy. OCT shows focal RPE thickening colocalized with hyper-autofluorescent lesions. Toxicity usually develops at cumulative doses over 1,000 g to 1,500 g, annual testing with OCT, NIR, and FAF as cumulative doses exceed 500 g.
Hydroxychloroquine and chloroquine toxicity: See 11.32, Chloroquine/Hydroxychloroquine Toxicity.
Syphilis: Positive bloodwork, asymmetric visual fields, abnormal fundus appearance, may have a history of recurrent uveitis. No family history of RP. The ERG is usually preserved to some degree.
Congenital rubella: A salt-and-pepper fundus appearance may be accompanied by microphthalmos, cataract, deafness, a congenital heart abnormality, or another systemic abnormality. The ERG is usually normal.
After resolution of a serous or RRD: The history is diagnostic (e.g., toxemia of pregnancy, Harada disease, chronic CSCR).
Pigmented paravenous retinochoroidal atrophy: Paravenous localization of RPE degeneration and pigment deposition. No definite hereditary pattern. Variable visual fields and ERG (usually normal).
After severe blunt trauma: Usually due to spontaneous resolution of RD.
Carriers of ocular albinism: See 13.8, Albinism.
Vitamin A deficiency: From malabsorption or poor diet. See 13.7, Vitamin A Deficiency/xerophthalmia.
The pigment abnormalities are at the level of the RPE with phenothiazine toxicity, syphilis, and congenital rubella. With resolved RD, the pigment is intraretinal. |
Medical and ocular history pertaining to the diseases discussed previously.
Family history with pedigree and genetic testing for diagnostic and counseling purposes (see above). Full list of available genetic tests at http://www.ncbi.nlm.nih.gov/gtr/tests.
Dark-adaption studies and ERG: May help distinguish stationary rod dysfunction (congenital stationary night blindness) from RP (a progressive disease).
Vitamin A levels in cases where the patient has had gastrointestinal surgery or has poor nutrition.
Consider syphilis testing (RPR or VDRL and FTA-ABS or treponemal-specific assay).
If the patient is male and inheritance pattern is unknown, examine his mother and perform an FAF on her. Female carriers of X-linked disease often have abnormal pigmentation in the midperiphery and abnormal results on FAF.
If neurologic abnormalities such as ataxia, polyneuropathy, deafness, or anosmia are present, obtain a fasting (at least 14 hours) serum phytanic acid level to rule out Refsum disease.
If hereditary abetalipoproteinemia is suspected, obtain serum cholesterol and triglyceride levels (levels are low), a serum protein and lipoprotein electrophoresis (lipoprotein deficiency is detected), and peripheral blood smears (acanthocytosis is seen).
If Kearns–Sayre syndrome is suspected, the patient must be examined by a cardiologist with sequential EKGs; patients can die of complete heart block. All family members should be evaluated.
For syphilis, see 12.10, Syphilis. For vitamin A deficiency, see 13.7, Vitamin A Deficiency/Xerophthalmia.
No definitive treatment for RP is currently known although gene therapy and other treatments are starting to emerge. See above, Retinitis Pigmentosa.
Cataract surgery may improve central visual acuity. Topical or oral carbonic anhydrase inhibitors (e.g., acetazolamide 500 mg/d) may be effective for CME.
All patients benefit from genetic counseling and instruction on how to deal with their visual handicaps. Tinted lenses may provide comfort outdoors and may provide better contrast enhancement. In advanced cases, low-vision aids and vocational rehabilitation are helpful.
HEREDITARY CHORIORETINAL DYSTROPHIES AND OTHER CAUSES OF NYCTALOPIA (NIGHT BLINDNESS)
Gyrate atrophy: Nyctalopia and decreased peripheral vision usually presenting in the first decade of life, followed by progressive constriction of visual field. Scalloped RPE and choriocapillaris atrophy in the midperiphery during childhood that coalesces to involve the entire fundus, posterior subcapsular cataract, high myopia with astigmatism. Constriction of visual fields and abnormal to nonrecordable ERG. Plasma ornithine level is 10 to 20 times normal; lysine is decreased. Consider genetic testing. Autosomal recessive.
Reduce dietary protein consumption and substitute artificially flavored solutions of essential amino acids without arginine (e.g., arginine-restricted diet). Monitor serum ammonia levels.
Supplemental vitamin B6 (pyridoxine). The dose is not currently established; consider 20 mg/d p.o. initially and increase up to 500 mg/d p.o. if there is no response. Follow serum ornithine levels to determine the amount of supplemental vitamin B6 and the degree to which dietary protein needs to be restricted. Serum ornithine levels between 0.15 and 0.2 mmol/L are optimal.
Choroideremia: Males present in the first to second decade of life with nyctalopia, followed by insidious loss of peripheral vision. Decreased central vision occurs late. In males, early findings include dispersed pigment granules in the periphery with focal areas of RPE atrophy. Late findings include total absence of RPE and choriocapillaris. No bone spicules. Retinal arteriolar narrowing and optic atrophy can occur late in the process. Constriction of visual fields, normal color vision, markedly reduced ERG. Female carriers have small, scattered, square intraretinal pigment granules overlying choroidal atrophy, most marked in the midperiphery. FAF is helpful, as well as genetic testing. No effective treatment for this condition is currently available. Darkly tinted sunglasses may ameliorate symptoms. X-linked recessive. Consider genetic counseling.
Vitamin A deficiency: Marked night blindness. Numerous small, yellow-white, well-demarcated spots deep in the retina seen peripherally. Dry eye and/or Bitôt spots (white keratinized lesions) on the conjunctiva. See 13.7, Vitamin A Deficiency/Xerophthalmia.
Zinc deficiency: May cause abnormal dark adaptation (zinc is needed for vitamin A metabolism).
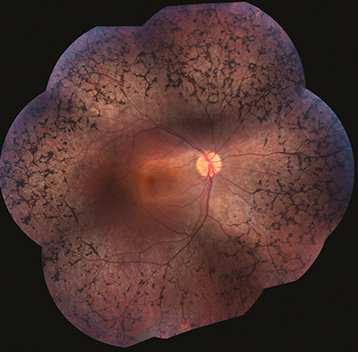
Congenital stationary night blindness: Night blindness from birth, normal visual fields; may have a normal or abnormal fundus, not progressive. Paradoxical pupillary response. One variant is Oguchi disease, characterized by the Mizuo–Nakamura phenomenon—the fundus has a tapetum appearance in the light-adapted state but appears normally colored when dark adapted (may take up to 12 hours) (See Figures 11.28.2 and 11.28.3).
Undercorrected myopia: The most common cause of poor night vision.