Decreased vision in childhood or young adulthood. Early in the disease, the decrease in vision is often out of proportion to the clinical ophthalmoscopic appearance; therefore, care must be taken not to label the child a malingerer.
(See Figures 11.30.1 to 11.30.3.)
Figure 11.30.1: Stargardt disease.
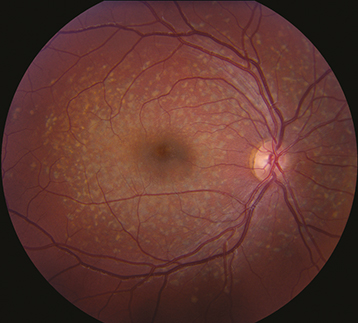
Figure 11.30.2: Intravenous fluorescein angiography of Stargardt disease exhibiting silent choroid.
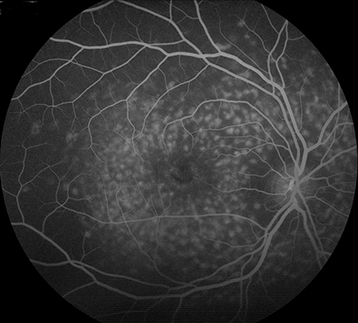
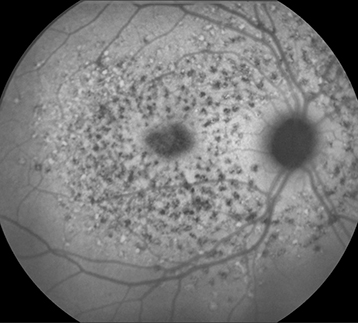
Figure 11.30.3: Fundus autofluorescence in Stargardt disease.
Critical
Any of the following may be present.
A relatively normal-appearing fundus except for slight granularity in the foveola.
Yellow or yellow-white, fleck-like deposits at the level of the RPE, often in a pisciform (fish-tail) configuration.
Atrophic macular degeneration: May have a bull’s eye appearance as a result of atrophy of the RPE around a normal central core of RPE, a “beaten-metal” appearance, pigment clumping, or marked GA.
|
 NOTE NOTEIn early stages, vision declines before visible macular changes develop. |
Other
Vermilion or light-brown fundus with obscuration of choroidal vasculature. Atrophy of the RPE just outside of the macula or in the midperipheral fundus, normal peripheral visual fields in most cases, and rarely an accompanying cone or rod dystrophy. Peripapillary sparing best seen by FAF. The ERG is typically normal in the early stages but may become abnormal late in the disease. The EOG can be subnormal.
Inheritance
Usually autosomal recessive, but occasionally autosomal dominant (dominant cases may be asymptomatic into middle age).
Indicated when the diagnosis is uncertain or must be confirmed.
History: Age at onset, medications, family history?
Dilated fundus examination.
OCT may show photoreceptor disorganization, outer retinal atrophy, and RPE changes, which may appear early even with a normal fundus examination.
IVFA often shows blockage of choroidal fluorescence producing a “silent choroid” or “midnight fundus” as a result of increased lipofuscin in RPE cells.
FAF can be helpful in diagnosis and in monitoring disease progression.
ERG and EOG.
Formal visual field examination (e.g., Octopus, Humphrey).
Consider genetic testing: Sequencing of the ABCA4 gene (found to be abnormal in many cases of Stargardt disease and other related maculopathies).
Avoid vitamin A supplementation. Ultraviolet light-blocking glasses when outdoors may be beneficial. Lutein and zeaxanthin supplementation might be helpful. Low-vision aids, services dedicated to helping the visually handicapped, and genetic counseling are helpful. Trials of vitamin A antagonists and gene therapy are ongoing. Safety glasses might be of help since trauma to the retina can cause an inflammatory response to the accumulating lipofuscin.