


A.3. What is the pathogenesis of aortic aneurysms and what genetic conditions predispose to their formation?
Answer:
TAAs primarily arise from cystic medial degeneration of the elastic and muscular tissue of the aortic wall. The resulting tissue weakness predisposes the wall to dilate under the stress of high pressures within the aorta. The dilation of the aorta is associated with decreased vessel wall compliance, increased wall stress, and a propensity for subsequent rupture of the aneurysm. Patients who develop TAAs are also predisposed to developing AAAs and cerebral aneurysms. Cystic medial degeneration occurs with aging but can be accelerated in male and in the presence of certain acquired risk factors, such as hypertension, smoking, atherosclerosis, and trauma, in addition to genetic, congenital, infectious, and autoimmune disease processes that are associated with aneurysm formation.
The presence of TAA in younger patients is often attributed to heritable causes, some of which are limited to the aorta while others have systemic features.
Marfan syndrome (MFS) is an autosomal dominant disorder caused by a mutation in the fibrillin-1 gene, which provides a framework for the stability and elasticity of the aorta. Patients with MFS have a characteristic overgrowth of the long bones of the arms and legs, increased finger length named arachnodactyly, and joint laxity. They can also demonstrate ocular abnormalities, most commonly lens dislocation. In addition, patients are at increased risk of aortic root dilation, ascending and descending aortic aneurysms, and aortic dissection. Atrioventricular valvular disease may occur, most commonly due to prolapse and regurgitation. Dilated cardiomyopathy and pulmonary hypertension associated with mitral regurgitation are thought to be the leading cause of MFS-related death in infancy. In addition to the 1847 fibrillin-1 mutations linked to MFS, increased transforming growth factor-β (TGF-β) signaling contributes to the increased risk of TAAs and dissections in MFS patients. Fibrillin-1 sequesters TGF-β within the walls of the aorta and therefore, fibrillin-1 mutations have been implicated in impaired TGF-β sequestering and homeostasis.
Vascular Ehlers-Danlos syndrome (vEDS), also known as Ehler-Danlos syndrome type IV, has an autosomal dominant inheritance and results from mutations in the collagen type III α 1 gene (COL3A1). Patients are predisposed to translucent and fragile skin, varicose veins, atrophic scars, fistula formation, and organ rupture, such as spontaneous intestinal or uterine perforation, in addition to the development of aneurysms and vessel dissections. Children can also present with an inguinal hernia, pneumothorax, or recurrent joint dislocations. Although diagnosis is often made clinically, many patients are only diagnosed after a major arterial or gastrointestinal event. In all, 80% of patients with vEDS experience a major complication prior to the age of 40 years. Clear genotype-phenotype associations are hindered by significant heterogeneity between patients with identical mutations.
Loeys-Dietz syndrome (LDS) is an autosomal dominant disease that has been linked with numerous mutations, such as TGFBR1, TGFBR2, SMAD3, and TGFB2, which correspond to LDS1-4 subtypes, respectively. Excessive collagen deposition in arterial walls due to increased TGFβ signaling results in dysfunctional elastogenesis, which leads to aortic and branch vessel aneurysms and dissections, arterial tortuosity, and intracranial aneurysms. LDS also manifests with characteristic hypertelorism, skeletal and craniofacial abnormalities, and translucent skin. There is substantial clinical variability within and between individuals with all four LDS subtypes, though LDS4 likely represents the mildest disease. Relative to patients with MFS, the clinical course of LDS1/2 patients appears to be more severe. Diagnosis is often made after a significant aortic event.
Bicuspid aortic valve (BAV) is the most common congenital cardiac abnormality and has an autosomal dominant inheritance. Accelerated aortic dilatation, which prevails in 50% of patients with BAV, commonly involves the aortic root and ascending aorta (Figure 9.5). Patients with BAV are 8 times more likely to have aortic dissections relative to the general population. The genetic basis for BAV aortopathy is complex and thought to be related to the loss of a dynamic balance between the expression of matrix metalloproteinases (MMPs) and endogenous inhibitors (matrix metalloproteinase organization inhibitors, TIMPs). For instance, mutations in TIMP3 are associated with a greater incidence of BAV and TAA in Turner syndrome. Turner syndrome is a genetic disorder characterized by the lack of an X-chromosome in a female patient (45, X). Females with Turner syndrome are often short in stature, have dysfunctional ovaries, and are at increased risk of cardiovascular disease. BAV disease is present in between 10% and 25% of females with Turner syndrome, and approximately 8% have coarctation of the aorta. To date, variants in 37 genes have been associated with TAA and dissection, yet up to 80% of familial TAAs have an unknown molecular etiology. Advances in genomic sequencing and bioinformatics tools will accelerate progress with the ultimate goal of providing personalized aortic care and tailoring surgical recommendations to individual genetic profiles.
Figure 9.5.: A. Midesophageal Aortic Valve (Av) Short-Axis View on Transesophageal Echocardiography Showing a Bicuspid Valve, with the Fusion of the Left and Right Coronary Cusps.
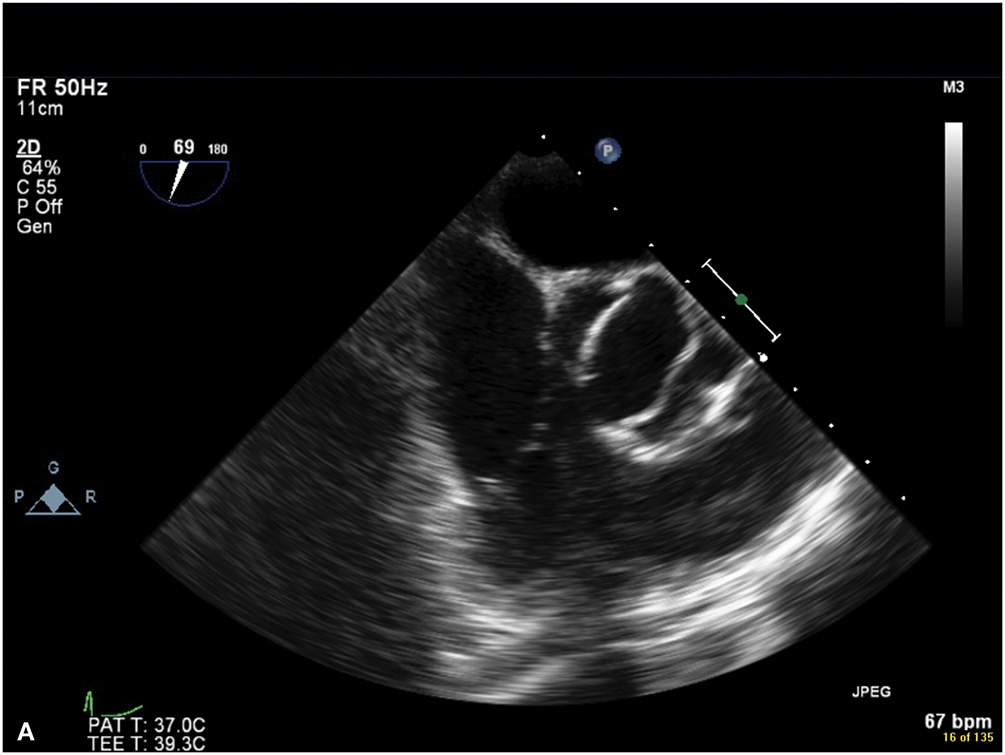
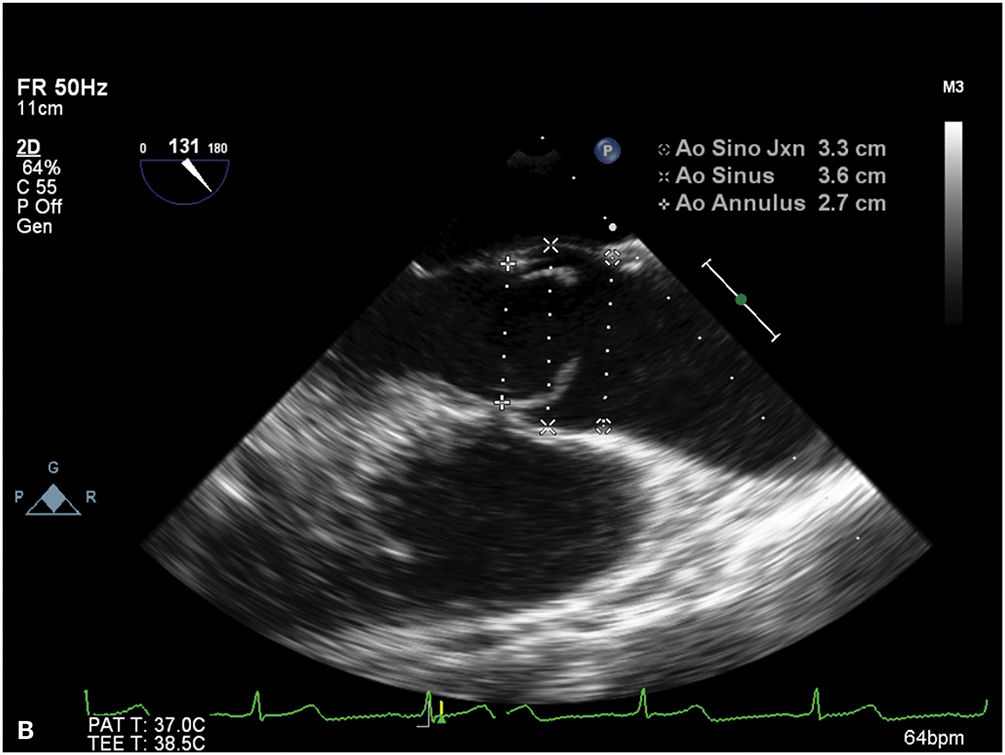
A. Midesophageal aortic valve (AV) short-axis view on transesophageal echocardiography showing a bicuspid valve, with the fusion of the left and right coronary cusps. B. Midesophageal aortic valve long-axis view on transesophageal echocardiography demonstrating the characteristic doming of a stenotic bicuspid AV and blunting of the sinotubular junction due to aortic root (ascending aortic) dilatation.
There are additional rheumatologic, infectious, and traumatic etiologies for TAA that merit consideration. Giant cell arteritis and Takayasu arteritis are the most common causes of aortitis. Aortic infections are most commonly caused by the spread of Staphylococcus aureus, Pneumococcus, Escherichia coli, and Salmonella via a vulnerable plaque or preexisting aneurysm. Syphilitic aortitis, which occurs 10 to 25 years after systemic Treponema pallidum infection, is rare with antibiotic treatment. Fungal aortitis from Candida or Aspergillus and tuberculous aortitis may be seen in immunocompromised patients. HIV infection is associated with an increased risk of TAA formation. Furthermore, trauma from deceleration injuries can also lead to TAAs, typically at the aortic isthmus.
References
- Faggion Vinholo T, Brownstein AJ, Ziganshin BA, et al. Genes associated with thoracic aortic aneurysm and dissection: 2019 update and clinical implications. Aorta (Stamford). 2019;7:99-107.
- Huguenard AL, Gupta VP, Braverman AC, Dacey RG. Genetic and heritable considerations in patients or families with both intracranial and extracranial aneurysms. J Neurosurg. 2021;134:1999-2006.
- Isselbacher EM, Preventza O, Hamilton Black J 3rd, et al; Peer Review Committee Members. 2022 ACC/AHA guideline for the diagnosis and management of aortic disease: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. Circulation. 2022;146:e334-e482.
- Jones JA, Ikonomidis JS. The pathogenesis of aortopathy in Marfan syndrome and related diseases. Curr Cardiol Rep. 2010;12:99-107.
- Ostberg NP, Zafar MA, Ziganshin BA, Elefteriades JA. The genetics of thoracic aortic aneurysms and dissection: a clinical perspective. Biomolecules. 2020;10:182.