Unilateral or bilateral congenital abnormalities of the cornea, iris, anterior chamber angle, and lens.
Most Common Entities
Microcornea: Horizontal corneal diameter small for age (<10 mm at age 2 years). May be isolated or associated with microphthalmia, cataract, or nanophthalmos.
Posterior embryotoxon: A prominent, anteriorly displaced Schwalbe line. Higher risk for the development of early-onset glaucoma. May be normal or seen in association with Axenfeld–Rieger and Alagille syndromes.
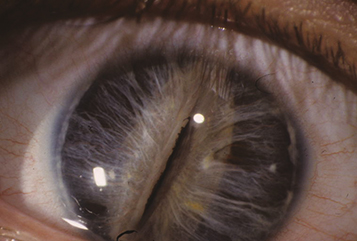
Axenfeld–Rieger spectrum: Ranges from posterior embryotoxon and iris strands inserting onto Schwalbe line or the cornea to more severe iris malformations including polycoria and corectopia. Glaucoma develops in 50% of patients. Heterozygous mutations in PITX2 and FOXC1 are most common. May be associated with abnormal teeth (e.g., microdontia, conical teeth, hypodontia), skeletal, cardiac valve and pituitary abnormalities, redundancy of the periumbilical skin, deafness, and intellectual disability (See Figure 8.14.1).
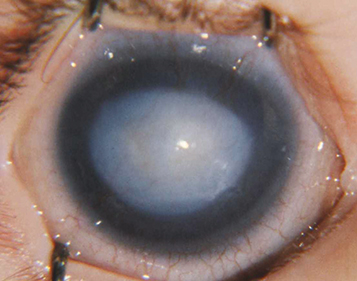
Peters anomaly: Central posterior corneal defect with opacity of overlying sclera, may have iris strands to defect (Peters type 1) or adherence of cataractous lens to defect (Peters type 2). Associated with many gene mutations including PAX6, CYP1B1. One-third with systemic abnormalities which warrants a full pediatric workup. “Peters plus” syndrome is characterized by an associated skeletal dysplasia with short stature and intellectual disability (See Figure 8.14.2).
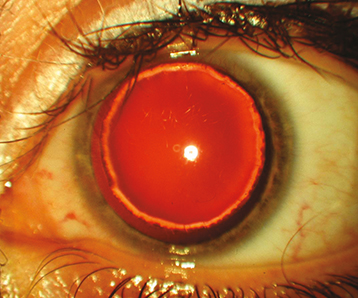
Microspherophakia: The lens is small and spherical in configuration. It can subluxate into the anterior chamber, causing a secondary glaucoma. Can be isolated or seen in association with Weill–Marchesani syndrome (See Figure 8.14.3).
Anterior and posterior lenticonus: An anterior or posterior ectasia of the lens surface, posterior occurring more commonly than anterior. Often associated with cataract. Unilateral or bilateral. Anterior lenticonus is associated with Alport syndrome. Posterior lenticonus is usually isolated but may be autosomal dominant, can also be seen with Lowe syndrome.
Ectopia lentis: See 13.2, Subluxed or Dislocated Crystalline Lens.
Ectopia lentis et pupillae: Lens displacement associated with pupillary displacement in the opposite direction. Usually not associated with glaucoma.
Aniridia: Bilateral, near-total absence of the iris. Glaucoma, macular hypoplasia, nystagmus, refractive error, and corneal pannus are common. Aniridia is autosomal dominant in two-thirds of patients, with small missense or nonsense mutations in PAX6. Especially in sporadic cases, if PAX6 is deleted as part of a larger chromosomal deletion, it is called WAGR syndrome (Wilms tumor, aniridia, genital abnormalities, intellectual disability). Children with sporadic aniridia have about a 30% chance of Wilms tumor development and require genetic screening as well as renal US.
Iris coloboma: failure of embryonic fissure closure. Typically present as inferonasal gap in the iris, but also can involve ciliary body, choroid, retina, and optic nerve.
Sclerocornea: Descriptive term applied to nonprogressive, congenital, partially or totally opaque cornea. Unilateral or bilateral. May be mild and peripheral or severe and diffuse. Associated with severe anterior segment dysgenesis, flat cornea, and risk for glaucoma.
Primary aphakia: Failure of lens development. Usually associated with microphthalmia and severe intraocular dysgenesis including retinal dysplasia and corneal opacity. High risk for glaucoma.