AUTHOR: Michael Z. Moore, MD


Neuromyelitis optica spectrum disorder (NMOSD) is an uncommon autoimmune inflammatory demyelination disorder of the central nervous system (CNS). It is a clinical syndrome defined by predilection to affect particular regions of the CNS: Specifically the optic nerve, spinal cord (typically with longitudinally extensive transverse myelitis), and some areas of the brain including periependymal regions (most strikingly the area postrema). The clinical syndrome is further subdivided into seropositive NMOSD (AQP4-IgG+ - 80% of patients) and seronegative NMOSD (MOG-IgG+, negative for both AQP4-IgG and MOG-IgG, or unknown serologic status).1
- Wide variety of estimated incidence, ranging from 0.037 per 100,000 in Australia and New Zealand to 0.73 per 100,000 in Afro-Caribbean region.2
Though presently not well understood, there has been postulated a genetic predisposition towards NMOSD. Specifically relevant to seropositive NMOSD, associations with AQP4-IgG seropositivity and HLA-DRB1∗03 (DR3) in French and Brazilian populations and HLA-DPB1∗0501 in Japanese and Chinese populations have been described.6
Findings depend on the location of the CNS lesion(s) and may include the following:
- Common: Nonspecific complaints such as fatigue (most common, with 80% lifetime prevalence), blurred vision, diplopia, vertigo, falls, hemiparesis, paraparesis, monoparesis, numbness, paresthesias, ataxia, cognitive impairment, depression, anxiety, pseudobulbar affect (involuntary crying or laughing out of context), sexual dysfunction, and bowel/bladder dysfunction
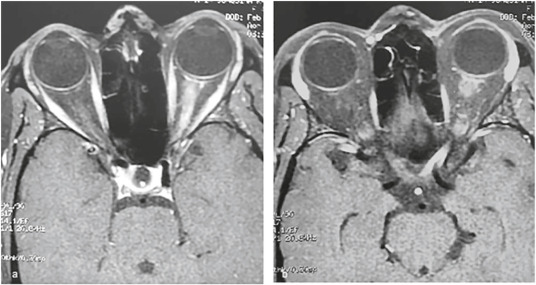
- Optic neuritis: Unilateral or bilateral
- Corticospinal tract(s) involvement: Transverse myelitis often involving three or more segments of the spinal cord, upper motor neuron signs such as spasticity (particularly leg spasms at night or after prolonged immobility), hyperreflexia, clonus, extensor plantar responses, tonic spasms, upper motor neuron pattern of weakness
- Area postrema syndrome: Intractable hiccupping and/or vomiting (occurs in up to 60% of patients during their disease course)
- Brainstem syndromes including combinations of cranial nerve palsies, weakness, sensory loss, and ataxia.
- Diencephalic syndromes: Narcolepsy, thalamic/hypothalamic-mediated dysfunction
- Sensory involvement: May include partial or full dermatomal loss of pain and temperature, loss of vibration (common) and position sense, temperature dysregulation, thoracic band of sensory loss, paresthesias, trigeminal neuralgia
- Bladder dysfunction: Due to spinal cord involvement.
As AQP4 is expressed in the central nervous system most abundantly on the foot processes of astrocytes, seropositive NMOSD should be understood as an antibody-mediated astrocytopathy. In a clinical relapse, accumulation of AQP4-IgG stimulates an inflammatory response mediated by such factors as complement activation and interleukins such as interleukin-6 (IL-6), leading to the influx of granulocytes. This cascades into a substantial local inflammatory response, leading to bystander damage of local cells and cell products such as myelin or axons themselves. On the other hand, MOG-IgG associated NMOSD is more clearly demyelinating, with some histopathologic studies bearing features similar to type II MS pattern demyelinating lesions.6 The specifics of non-MOG-IgG seronegative NMOSD pathophysiology are less well understood.