Workup
- History: Determine the patient’s age and rapidity of onset of the visual loss. Previous episode? Pain with eye movement? Intractable hiccups, nausea/vomiting, and severe itching are features strongly suggestive of NMOSD.
- Complete ophthalmic and neurologic examinations, including pupillary and color vision assessment, evaluation for vitreous cells, and dilated retinal examination with attention to the optic nerve.
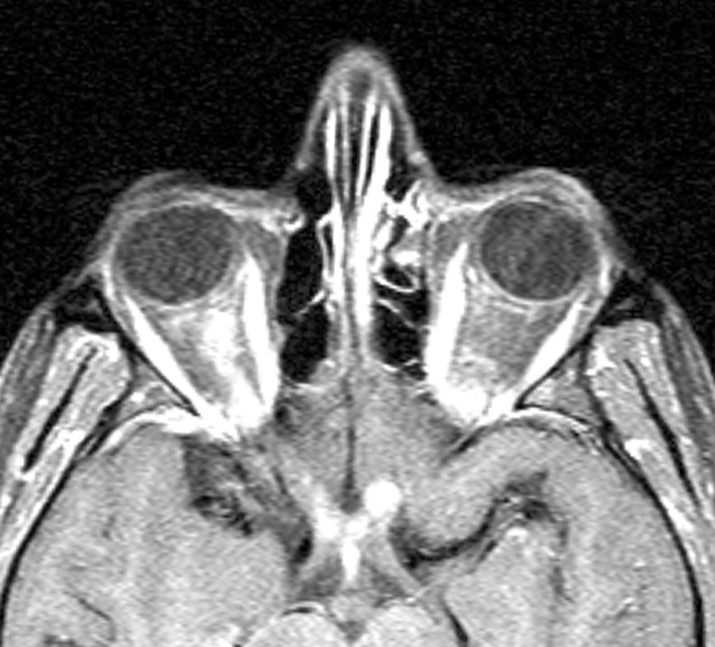
- For all cases, MRI of the brain and orbits with gadolinium and fat suppression should be obtained (see Figure 10.14.1). While a short segment of optic nerve is involved in typical optic neuritis, lesions in NMOSD may be longitudinally extensive. In addition, patients with NMOSD should have MRI of the spine to look for signs of transverse myelitis. Antibodies for anti-aquaporin 4 (anti-AQP4) and anti-myelin oligodendrocyte glycoprotein (anti-MOG) should be drawn.
- Check blood pressure.
- Visual field test, preferably automated (e.g., Humphrey).
- Consider the following: CBC, ESR, anti-AQP4 and anti-MOG antibodies for NMOSD, ACE level, Lyme antibody, FTA-ABS or treponemal-specific assay and RPR or VDRL tests, and chest x-ray or CT.
 NOTE:
NOTE: