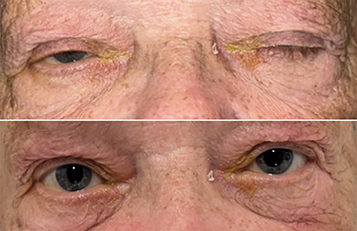
Eaton–Lambert syndrome: A myasthenia-like paraneoplastic condition associated with carcinoma, especially lung cancer. Isolated eye signs do not occur, although eye signs may accompany systemic signs of weakness. Unlike myasthenia, muscle strength increases after exercise. Electromyography (EMG) distinguishes between the two conditions.
Myasthenia-like syndrome due to medication (e.g., penicillamine, aminoglycosides).
CPEO: No diurnal variation of symptoms or relation to fatigue; usually a negative intravenous edrophonium chloride test. Typically no diplopia. See 10.12, Chronic Progressive External Ophthalmoplegia.
Kearns–Sayre syndrome: CPEO and retinal pigmentary degeneration in a young person; heart block develops. See 10.12, Chronic Progressive External Ophthalmoplegia.
Third cranial nerve palsy: Pupil may be involved, no orbicularis weakness, no fatigability, no diurnal variation. See 10.5, Isolated Third Cranial Nerve Palsy.
Horner syndrome: Miosis accompanies the ptosis. Pupil does not dilate well in darkness. See 10.2, Horner Syndrome.
Levator muscle dehiscence or disinsertion: High eyelid crease on the side of the droopy eyelid, no variability of eyelid droop, no orbicularis weakness.
Thyroid eye disease: No ptosis. May have eyelid retraction or eyelid lag, may or may not have exophthalmos, no diurnal variation of diplopia. Graves disease occurs in 5% of patients with myasthenia gravis. See 7.2.1, Thyroid Eye Disease.
Idiopathic orbital inflammatory syndrome: Proptosis, pain, ocular injection. See 7.2.2, Idiopathic Orbital Inflammatory Syndrome.
Myotonic dystrophy: May have ptosis and rarely, gaze restriction. After a handshake, these patients are often unable to release their grip (myotonia). Polychromatic lenticular deposits (“Christmas tree” cataract) and pigmentary retinopathy present.