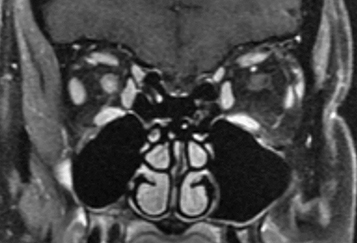
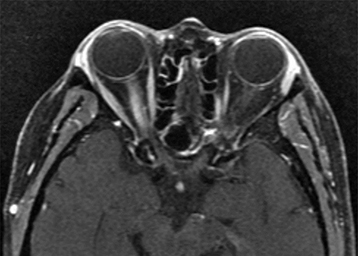
Typical optic neuritis associated with MS causes vision loss over hours to days, with the nadir approximately 1 week after onset. Visual loss may be subtle or profound. Usually unilateral, rarely bilateral. Age typically 18 to 45 years. Retro-orbital pain, especially with eye movement. Acquired loss of color vision. Reduced perception of light intensity. May have other focal neurologic symptoms (e.g., weakness, numbness, tingling in extremities). May have antecedent flu-like viral syndrome. Occasionally altered perception of moving objects (Pulfrich phenomenon) or a worsening of symptoms with exercise or increase in body temperature (Uhthoff sign).
More recently, immune-mediated disorders of the CNS, such as neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD), have been described as causes of atypical optic neuritis. Atypical features include advanced patient age, severe vision loss with poor recovery, and simultaneous or rapidly sequential bilateral optic neuritis.
Critical
Relative afferent pupillary defect in unilateral or asymmetric cases; decreased color vision; central, cecocentral, or arcuate visual field defects.
Other
Swollen disc (in one-third of patients) usually without peripapillary hemorrhages (papillitis most commonly seen in children and young adults) or a normal disc (in two-thirds of patients; retrobulbar optic neuritis more common in adults). Posterior vitreous cells possible.
Typical Optic Neuritis
If patient seen acutely with no prior history of MS:
Offer pulsed i.v. steroid in the following regimen within 14 days of decreased vision:
Methylprednisolone 1 g/d i.v. for 3 days, then
Prednisone 1 mg/kg/d p.o. for 11 days, then
Taper prednisone over 4 days (20 mg on day 1, 10 mg on days 2 through 4).
Antiulcer medication (e.g., omeprazole 20 mg p.o. daily or ranitidine 150 mg p.o. b.i.d.) for gastric prophylaxis.
|
 NOTE NOTEThe Optic Neuritis Treatment Trial (ONTT) found steroid treatment reduced initial progression to clinically definite multiple sclerosis (CDMS) for 2 years. Steroid therapy only increases the rapidity of visual return but does not improve final visual outcome. |
If MRI shows two or more characteristic demyelinating lesions, treat with the aforementioned steroid regimen and refer to neurologist or neuro-ophthalmologist for further management. If MS is suspected, treatment may be initiated with disease-modifying therapies. These include agents that are given orally, by injection, or by infusion. The most commonly used medications include interferon-beta, glatiramer acetate, fingolimod, dimethyl fumarate, teriflunomide, alemtuzumab, natalizumab, ocrelizumab, and dalfampridine.
Patients with one or more typical signal changes on MRI have a 72% chance of developing CDMS over 15 years.
|
| NOTE NEVER use oral prednisone as a primary treatment because of increased risk of recurrence found in ONTT. Disease-modifying drugs as listed above have been shown to reduce probability of progression to CDMS in high-risk patients. |
With a negative MRI, the risk of MS is low, 25% at 15 years. Thus, observation was an acceptable option in the past. However, in the current era of NMOSD, negative MRI should arouse suspicion for this condition. Pulsed i.v. steroid should be administered in all patients, and additional serologic studies for antibodies should be obtained.
In a patient with a diagnosis of prior MS or typical optic neuritis:
Observation or pulsed i.v. steroids, as above. We usually treat with i.v. pulsed steroids.
Atypical Optic Neuritis
Anti-AQP4 positive NMOSD, anti-MOG positive MOGAD, or seronegative disease:
For acute optic neuritis, high-dose i.v. steroids are used first.
Plasmapheresis should be performed if response to steroids is poor.
Referral to a neurologist/neuroimmunologist for long-term immunosuppression with agents such as rituximab, azathioprine, or mycophenolate. Food and Drug Administration (FDA)-approved medications for the treatment of NMOSD include inebilizumab, satralizumab, and eculizumab.
Diagnosis of Prior MS or Typical Optic Neuritis
Reexamine the patient approximately 4 to 6 weeks after presentation and then every 3 to 6 months.
Clinically isolated syndromes without features of MS need repeat MRI brain every 6 months initially and then annually to monitor for development of MS lesions.
Patients at high risk for MS, including patients with CNS demyelination on MRI or a positive neurologic examination, should be referred to a neurologist or neuro-ophthalmologist for evaluation and management of possible MS.
Atypical Optic Neuritis
NMOSD: Closer follow-up initially may be necessary due to frequency of relapses and severity of vision loss. Follow-up within 2 weeks of initial treatment with rapid referral to neurologist experienced in treating NMOSD.
BeckRW, ClearyPA, AndersonMMJr, et al.A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med. 1992;326:581–588.
BeckRW, ClearyPA, TrobeJD, et al.The effect of corticosteroids for acute optic neuritis on the subsequent development of multiple sclerosis. The Optic Neuritis Study Group. N Engl J Med. 1993;329:1764–1769.
ChenJ, PittockS, FlanaganE, et al.Optic neuritis in the era of biomarkers. Surv Ophthalmol. 2020;65(1):12–17.
JacobsLD, BeckRW, SimonJH, et al.Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Engl J Med. 2000;343:898–904.
Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final follow-up from the Optic Neuritis Treatment Trial. Arch Neurol. 2008;65:727–732.