Decreased vision, photophobia, pain, and red eyes; accompanied or preceded by a headache, stiff neck, nausea, vomiting, fever, and malaise. Hearing loss, noise causing ear pain, and tinnitus frequently occur. Typically bilateral.
Autoimmune disease targeted against melanocyte-containing tissues resulting in bilateral granulomatous panuveitis with skin, meningeal, and auditory-vestibular involvement.
Decreased vision, photophobia, pain, and red eyes; accompanied or preceded by a headache, stiff neck, nausea, vomiting, fever, and malaise. Hearing loss, noise causing ear pain, and tinnitus frequently occur. Typically bilateral.
Diagnostic criteria include the following:
(1) No history of ocular trauma, (2) no evidence of other disease processes, (3) bilateral anterior or panuveitis, (4) neurologic/auditory findings (usually occur before ocular disease), and (5) dermatologic findings (usually occur after ocular disease).
Complete VKH includes criteria 1 to 5, incomplete VKH includes criteria 1 to 3 and either 4 or 5, and probable VKH (isolated ocular disease) includes criteria 1 to 3.
Anterior: AC flare and cells, granulomatous (mutton fat) KP, perilimbal vitiligo (e.g., depigmentation around the limbus).
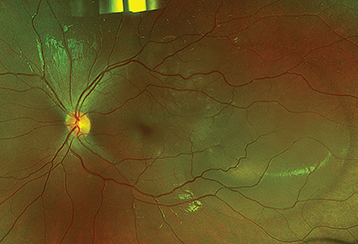
Posterior: Bilateral serous retinal detachments with underlying choroidal thickening, vitreous cells, opacities, and optic disc edema (see Figure 12.7.1).
Prodromal: Loss of high-frequency hearing, tinnitus, meningismus, encephalopathy, and hypersensitivity of the skin to touch.
Convalescent: Alopecia, vitiligo, poliosis, “sunset glow” fundus (yellow-orange appearance of the fundus due to depigmentation of the RPE and choroid).
Chronic recurrent: Recurrence of anterior uveitis, subretinal fibrosis, neovascularization, glaucoma, and cataract.
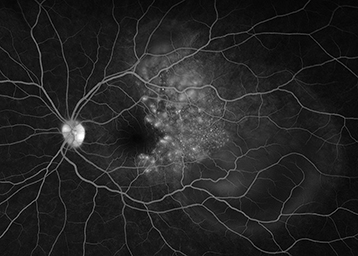
IVFA: Multiple pinpoint leaking areas of hyperfluorescence at the level of the RPE (see Figure 12.7.2).
Anterior: Iris nodules, peripheral anterior or posterior synechiae, hypotony, or increased IOP from the forward rotation of ciliary processes.
Posterior: Sunset glow fundus (mottling and atrophy of the RPE after the serous retinal detachment resolves), associated retinal vasculitis, CNV, and chorioretinal scars.
Systemic: Neurologic signs, including loss of consciousness, paralysis, and seizures.
Epidemiology
Typically, patients are aged 20 to 50 years, female (77%), and have pigmented skin (especially Asian, Middle Eastern, Hispanic, or Native American).
See Table 12.7.1 for the differential diagnosis of serous retinal detachments and 12.3, Posterior Uveitis. In particular, consider the following:
Sympathetic ophthalmia: History of trauma or surgery (especially repeated vitreoretinal procedures) in the contralateral eye. Usually no systemic signs.
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE): Ophthalmoscopic and IVFA features may be very similar, but there is less vitreous inflammation and no anterior segment involvement. See 12.8, White Dot Syndromes.
Posterior scleritis: Typically unilateral, not typically associated with neurologic or dermatologic findings. Associated with a “T” sign on B-scan ultrasound (US).
Systemic arterial hypertension and pregnancy-related hypertension: Acute elevation in blood pressure can produce multifocal serous retinal detachments.
Other granulomatous panuveitides (e.g., syphilis, sarcoidosis, and TB).
TABLE 12.7.1: Differential Diagnosis of Serous Retinal Detachments
| Central serous chorioretinopathy |
| Choroidal neovascularization |
| Choroidal tumors (including metastases) |
| Congenital optic disc pit |
| Disseminated intravascular coagulopathy |
| Eclampsia |
| Hyperviscosity syndrome |
| Hypotony |
| Uveal effusion syndrome |
| Malignant hypertension |
| Nanophthalmos |
| Posterior scleritis |
| Posterior uveitis |
| Retinal macroaneurysm |
| Retinal vein occlusion |
| Sympathetic ophthalmia |
| Toxemia of pregnancy |
| Vogt–Koyanagi–Harada syndrome |
See 12.4, Panuveitis, for a complete discussion on workup for patients with suspected VKH.
History: Neurologic symptoms, hearing loss, or hair loss? Previous eye surgery or trauma?
Complete ocular examination, including an IOP check and a dilated fundus examination.
Consider IVFA to evaluate for multifocal pinpoint leaking areas of hyperfluorescence at the level of the RPE and to track response to therapy.
Focused serologic testing based on history and examination:
Treponemal test (syphilis EIA, FTA-ABS, TP-PA), followed by confirmatory nontreponemal test (RPR, VDRL). See 12.10, Syphilis.
Interferon gamma releasing assay (IGRA, QuantiFERON Gold) and/or PPD. Consider chest imaging (CXR or CT chest) to assess for signs of active or prior pulmonary disease.
Consider B-scan US to assess for T-sign to rule out posterior scleritis.
Consider a CT or MRI of the brain with and without contrast in the presence of neurologic signs to rule out other CNS disease.
Lumbar puncture during attacks with meningeal symptoms for cell count and differential, protein, glucose, VDRL, Gram and methenamine–silver stains, and culture. Cerebrospinal fluid (CSF) pleocytosis is often seen in VKH and APMPPE.
Inflammation is initially controlled with steroids; the dose depends on the severity of the inflammation. In moderate to severe cases, the following regimen can be used. Steroids are tapered very slowly as the condition improves.
Topical steroids (e.g., prednisolone acetate 1% q.i.d. to q1h) while awaiting workup and to treat AC cells.
Topical cycloplegic (e.g., cyclopentolate 1% t.i.d. or atropine 1% b.i.d.).
Consider a trial of systemic steroids after appropriate negative infectious workup (e.g., prednisone 60 to 80 mg p.o. daily or intravenous methylprednisolone 1 g daily for 3 days followed by oral therapy) with concurrent calcium/vitamin D supplementation and antiulcer prophylaxis.
Consider local steroid injection, particularly if the disease is unilateral, the eye is pseudophakic, and there is no history of steroid response ocular hypertension.
If the disease becomes persistent consider long-acting local steroids (0.19 mg or 0.59 mg fluocinolone acetonide implants) versus systemic steroid-sparing immunosuppression (e.g., antimetabolites, calcineurin inhibitors, and anti-TNF factor agents).
Initial management may require hospitalization if intravenous corticosteroids are initiated.
Weekly, then monthly reexamination is performed, watching for recurrent inflammation and increased IOP.
Steroids are tapered very slowly, and most patients should be transitioned to steroid-sparing immunosuppressants for long-term management if the disease becomes persistent or incompletely responsive, or the patient is intolerant to systemic steroid therapy. Inflammation may recur up to 9 months after the steroids have been discontinued. If this occurs, steroids should be reinstituted.