AUTHORS: Harikrashna B. Bhatt, MD and Russell E. Bratman, MD



Definition
Multiple endocrine neoplasia (MEN) refers to a group of heritable genetic syndromes characterized by the development of specific groups of tumors of the endocrine glands.
Synonyms
| ||||||||||||||||||||||||
Physical Findings & Clinical Presentation
- Patients may present due to screening or may present with a MEN-associated tumor. Tumors are found incidentally due to biochemical abnormalities or to symptoms.
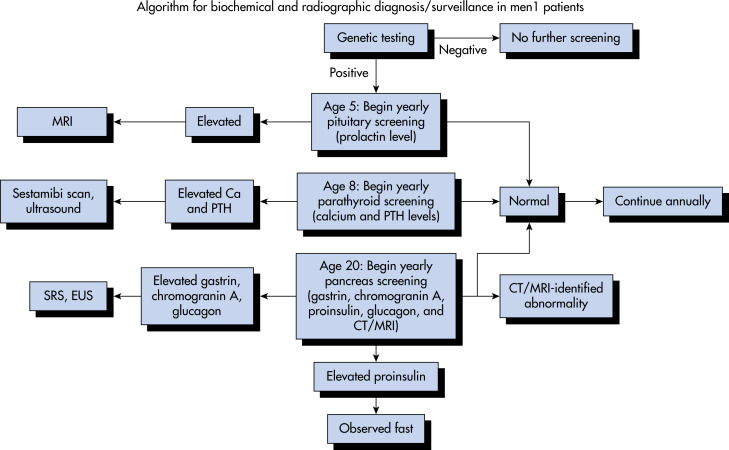
- MEN I (PPP [pituitary, pancreas, parathyroid])3,4
- Diagnostic criteria generally include two MEN-associated tumors, one tumor in a patient with a family history, or positive genetic testing
- Primary hyperparathyroidism (parathyroid adenoma or hyperplasia) is the most common manifestation and can cause hypercalcemia (urolithiasis, GI disturbance, bone pain, neuropsychiatric disturbances) and also affect bone density
- Pancreatic neuroendocrine tumors cause symptoms related to their secretory properties or metastases
- Gastrinoma: Peptic ulcers, diarrhea, esophageal symptoms (see “Gastrinoma”)
- Insulinoma: Hypoglycemic symptoms upon fasting and after exercise (see “Insulinoma”)
- Glucagonoma: Necrolytic migratory erythema (a blistering skin lesion), diabetes/glucose intolerance, weight loss
- VIPoma: Diarrhea, hypokalemia, decreased gastric acid
- Somatostatinoma: Hyperglycemia, cholelithiasis, diarrhea, abdominal pain, weight loss
- Nonfunctioning tumors can metastasize (frequently to the liver), a frequent cause of death in MEN I
- Pituitary tumors may cause compressive symptoms, such as visual field defects or hypopituitarism (see “Pituitary Adenoma”). Hormonal secretion may also cause symptoms.
- Prolactinoma (most common pituitary tumor in MEN I): Menstrual irregularities, hypogonadism, gynecomastia, and/or galactorrhea (see “Prolactinoma”)
- Somatotroph adenomas (producing growth hormone): Gigantism or acromegaly (frontal bossing, increased shoe/hat size, hyperglycemia, hyperhidrosis) depending on patient age (see “Acromegaly”)
- Corticotroph adenomas (producing adrenocorticotropic hormone): Cushingoid features such as weight gain, moon facies, hyperglycemia, bone loss, proximal muscle weakness, hypertension, hypokalemia (see “Cushing Disease and Syndrome”)
- Nonfunctioning (nonsecretory) tumors
- Other manifestations: Carcinoid tumors, collagenomas, angiofibromas, meningiomas, lipomas
- MEN IIA:5,6
- Medullary thyroid carcinoma (MTC) is a tumor of the thyroid’s calcitonin-secreting C cells. It can present with a neck mass, as well as flushing and diarrhea due to elevated calcitonin levels.
- Primary hyperparathyroidism: See “MEN I.” Less aggressive in “MEN IIA.”
- Pheochromocytoma is an adrenal tumor producing catecholamines, which can lead to life-threatening hypertensive crises.
- MEN IIB:5,6
Etiology
Tumor development facilitated by the previously described genetic mutations (Table E1).
TABLE E1 Tumors in Multiple Endocrine Neoplasia Syndromes
| Type (Chromosomal Location) | Tumors (Estimated Penetrance) | ||
|---|---|---|---|
| MEN 1 (11q13) | Parathyroid adenoma (90%) | ||
| Enteropancreatic tumor (30%-70%) Gastrinoma (40%) Insulinoma (10%) Nonfunctioning and PPoma (20%-55%) Glucagonoma (<1%) VIPoma (<1%) | |||
| Pituitary adenoma (30%-40%) Prolactinoma (20%) Somatotropinoma (10%) Corticotropinoma (<5%) Nonfunctioning (<5%) | |||
| Associated Tumors Adrenal cortical tumor (40%) Pheochromocytoma (<1%) Bronchopulmonary NET (2%) Thymic NET (2%) Gastric NET (10%) Lipomas (30%) Angiofibromas (85%) Collagenomas (70%) Meningiomas (8%) | |||
| MEN 2 (10 cen- 10q11.2) | |||
| MEN 2A | MTC (90%) Pheochromocytoma (50%) Parathyroid adenoma (20%-30%) | ||
| MTC only | MTC (100%) | ||
| MEN 2B (also known as MEN 3) | MTC (>90%) Pheochromocytoma Associated abnormalities (40%-50%) Mucosal neuromas Marfanoid habitus Medullated cornel nerve fibers Megacolon |
MEN, Multiple endocrine neoplasia; MTC, medullary thyroid carcinoma; NET, neuroendocrine tumor; PPoma, pancreatic polypeptidoma; VIPoma, vasoactive intestinal peptidoma.
Modified from Thakker RV: Multiple endocrine neoplasia type 1. In Jameson JL et al (eds): Endocrinology: adult and pediatric, Philadelphia, 2016, Saunders.